Введение
21,2 Производители интерлейкина (IL) -12 требуется для IFN-γ производство
21,3 Производители IFN-γ
Участие других цитокинов и
Регуляторные молекулы в сопротивлении 21.5 Вовлечение гуморального иммунитета в
Сопротивление
21,6 IFN-γиндуцированные эффекторные механизмы
Эффекторных клеток в головном мозге с активностью против T. гондия
Роль клеток, несущих Т. Gondii в мозге
Гены, участвующие в 21.9Host регулирования сопротивления
Генетические факторы T.gondii при определении развития Toxoplasma энцефалита и вирулентности
Иммунные эффекторные механизмы в глазной токсоплазмоз
Иммунные эффекторные механизмы в врожденном токсоплазмозе
Выводы
Подтверждения
Рекомендации
ВВЕДЕНИЕ

токсоплазма вездесущий, облигатное внутриклеточный паразит простейших у людей и животных. Хроническая (скрытая) инфекция с этим паразитом, вероятно, одна из самых распространенных инфекций человека, влияя на 10-25 процентов населения мира. Во время острой стадии инфекции, тахизоиты быстро пролиферируют в различные клетки и дра распространились по всей ткани хозяина. После острой стадии, паразит образует цисты (латентная стадия) в различных органах, особенно головной мозг, сердце,
и скелетных мышц, создание хронических Infec Тион. Инфекция у иммунокомпетентных лиц, как правило, незаметно или доброкачественная, самоограничение болезни, и приводит к скрытой хронической инфекции. Иммуносупрессия в хронически инфицированных инди-ческих лиц может привести к реактивации латентной Infec-ции, который инициируется нарушением кист, с последующим распространением тахизоитов. Такой REAC-tivation Т. гондия инфекции обычно представляет как токсоплазматический энцефалит (TE). TE стала крупной оппортунистических инфекционных заболеваний центральной нервной системы у больных СПИДом.
Токсоплазма. Модельные Apicomplexan-перспективы и методы, под редакцией Weiss & Kim
ISBN-13: 978-0-12-369542-0
ISBN-10: 0-12-369542-2
Copyright © 2007 Elsevier Ltd.
Все права воспроизведения в любой форме зарезервированы.

568 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
Так как иммунокомпетентные лица, как правило, не страдают очевидные неблагоприятные эффектами, ВКЛЮЧАЕТ-ки развития энцефалита, то ясно, что иммунный ответ является критическим для профилактики ТЭ. Паразит может также быть приобретен врожденно, и в этих случаях мозг является основным органом, пострадавших от инфекции.
Мышиные модели в основном были использованы для анализа механизмов резистентности хозяина к острой инфекции и развитию TE во время поздней стадии инфекции. IFN-γ-зависимый клеточный иммунитет был показан, играет важную роль в устойчивости к Т. гондиям, хотя гуморальный иммунитет также принимают участие. Ряд различных типов клеток, которые участвуют в сопротивлении в качестве производителей IFN-γ, Микроглии, которые находятся в головном мозге были выявлены, что популяция клеток, которая производит IFN-γпосле заражения с Т. гондиями (Suzuki и др., 2005). Несколько типов клеток участвуют также в качестве эффекторных клеток, которые становятся активированными IFN-γконтролировать паразит. Кроме того, несколько типов клеток, также важны как производители IL-12, который необходим для индукции IFN-γ,
21.2 ПРОДЮСЕРЫ интерлейкина (IL) -12 НЕОБХОДИМЫЕ ДЛЯ IFN-г ПРОИЗВОДСТВО
Дендритные клетки
IL-12 является наиболее важным индуктором ИФНγСинтез в острой стадии инфекции. Нейтрализация IL-12 с помощью анти-IL-12 антитела приводит к 100-процентной смертности у мышей, инфицированных с авирулентного штамма T. гондий и смертности связано со снижением ИФНγпроизводство (Gazzinelli и др., 1994). Дендритные клетки были идентифицированы как источник IL-12 в селезенке в ответ на Т. гондий (Reis е Соуза и др., 1997). Все ИЛ-12-позитивных клеток в селезенке Т. гондий-стимулированных мышей были обнаружены в зонах Т-клеток и были CD8α +, CD11c+, DEC205+дендритные клетки. CCR5, экспрессируется на поверхности дендритных клеток отвечают за их миграцию в селезеночные области Т-клетки после стимуляции (Reis электронного Соуза и др., 1997). сигнализации CCR5, также играет важную роль в активации
дендритные клетки для получения IL-12 (Aliberti и др., 2000). CCR5-дефицитный мышей имеет нарушенную продукцию IL-12 дендритные клетки, а также очень восприимчивы к инфекции Т. гондий. Циклофилин-18 была определена в качестве основной молекулы, полученной из паразита, который вызывает продукцию IL-12 через CCR5 (Aliberti и др., 2003). В последнее время профилин-подобный белок паразита было также установлено, что связываться с Toll-подобный рецептор 11 и стимулируют выработку IL-12 дендритными клетками (Яровинский и др., 2005).
В хронической стадии инфекции, клетки несут-ную дендритных клеточных маркеров, таких как CD11c и 33D1 расположены на участках воспаления в головном мозге мышей (Фишер и др., 2000). Эти мозга дендритные клетки созревают, как показано на высоком уровне Expres-Сьон молекул МНС класса II, CD40, CD54, CD80, CD86 и, и способны вызвать антиген-специфических Т-клеточного ответа в пробирке. Эти дендритные клетки, как было показано, что основными производителями IL-12 в мононуклеарных клетках, выделенных из мозга зараженных животных (Фишер и др., 2000). GM-CSF, по-видимому, важным для индукции этих дендритных клеток в культурах первичных клеток головного мозга с Т. гондий (Фишер и др., 2000). Так как IL-12 является необхо-сары для поддержания IFN-γ Производство в Т-клетках, опосредующих устойчивость к хронической инфекции (Яп и др., 2000), дендритные клетки в головном мозге зараженных мышей, вероятно, играют определенную роль в поддержании ИФНγ Производство Т-клеток в этом органе.
Макрофаги
Макрофаги производят IL-12 в ответ на тахи- zoites или тахизоит антигенов в пробирке (Gazzinelli и др., 1994, 1996). Так как макрофаги пропитать мозг мышей после заражения Т. гондий (Suzuki и др, 2005;.. Wilson и др, 2005), эти клетки, в дополнение к дендритным клеткам, может быть важным источником IL-12 в сопротивлении паразит в головном мозге.
Нейтрофилы
Нейтрофилы быстро проникают в брюшную полость мышей после внутрибрюшинного заражения Т. гондий. Приблизительно 85 процентов нейтрофилов отображается внутриклеточное хранение IL-12 (Bliss и др., 2000). Истощение нейтрофилов во время
| ПРОИЗВОДИТЕЛИ IFN-γ | 569 |
первые 6 дней инфекции привели к увеличению смертности у мышей в ассоциации с снижением продукции IL-12 и IFN- &γспленоцитами (Bliss и др., 2001). Быстрое проникновение нейтрофилов в очаг инфекции, как представляется, играет важ-Tant роли в индукции защитных иммунных реакций Th1-типа против Т. гондий на ранней стадии инфекции. Однако, неизвестно, будет ли нейтрофилы участвуют в сопротивлении в мозге, чтобы управлять Т. Gondii в хронической стадии инфекции.
21.3 ПРОИЗВОДИТЕЛИ IFN-γ
УЧАСТИЕ
Лимфотоксин
Лимфотоксин (LT), в дополнение к TNF-α, Является лиганд TNF R1. Шлютер и др. (2003) сообщили, что у мышей с дефицитом в LT не справился с управлением intracere-bral Т. Gondii и поддался некротический TE после заражения. IFN-γвыражение в их мозге было эквивалентно контрольным мышей, когда дефицитные животные было разработано TE. Эксперименты с костного мозга химеры мышах показали, что кроветворные клетки должны выразить как LT и TNF-α контролировать T.gondii, в головном мозге.
IL-4
CD4+Т-клетки, как известно, являются гетерогенными (Th1 и Th2) в отношении секреции цитокинов. Th1 клетки секретируют преимущественно IL-2 и IFN- &γ, Тогда как Th2 клетки предпочтительно производят ИЛ-4, ИЛ-5, ИЛ-6 и ИЛ-10. ИЛ-4, как сообщалось, имеет доминирующее влияние на определение паттерна цитокинов (Th2-типа), полученные CD4+Т-клетки при subse-дующей антигенной стимуляции в пробирке. Поскольку роль IFN-γимеет решающее значение для предотвращения развития ТЭ, роль IL-4 в иммунопатогенезе токсоплазмоза была адресована на мышах. Удивительно, IL-4-дефицитной (IL-4-/-) Мышей демон-Strate увеличение смертности по сравнению с контрольными животными (Roberts и др, 1996;.. Сузуки и др, 1996a;. Александр и др, 1998). Таким образом, ИЛ-4 играет защитную роль в устойчивости против Т. гондий. Следует отметить, однако, что сроки смертности и развития ТЭ в IL-4-/- является спорным.
Сузуки и др. (1996a) сообщили, что IL-4-/-мышей умерли во время поздней стадии (от 6 до 20 недель) инфекции, тогда как контрольные мыши все выжили. Смертность IL-4-/-мышей был связан с большими числами кист и областей острого очагового воспаления с тахизоитами в их головном мозге (Suzuki и др., 1996a). Эти результаты свидетельствуют о том, что IL-4 защищает от развития ТЭ, предотвращая образование кист и распространение
тахизоиты в головном мозге. В этих исследованиях, через 8 недель после заражения, клетки селезенки мышей контрольной группы получают значительно большее количество IFN-γ после стимуляции в пробирке с растворимыми антигенами Т. гондий, чем те, ИЛ-4-/-мышей (Сузуки и др., 1996а). Таким образом, IL-4, кажется, играет роль путем усиления IFN-γ производство во время поздней стадии инфекции, а также нарушение способности ИЛ-4-/- мыши, чтобы произвести ИФНγвероятно, способствует их восприимчивости к развитию тяжелой TE (Suzuki и др., 1996a). В связи с этими выводами, сообщалось, что IL-4 усиливает ИФНγ Produc-ции Т-клетки, которые уже были загрунтованы (дифференцированным), в то время как он подавляет дифференциальный-Тион нештрихованных Т-клеток IFN-γ-продуцирующих клетки (Благородный и Кемени, 1995). Во время инфекции с Т. гондий, IFN- &γ Производство происходит раньше, чем производство IL-4 (Бимэн и др., 1993). Таким образом, это может быть, что ИЛ-4 не влияет на дифференцировку нештрихованных Т-клеток к ИФНγ-продуцирующих клетки следующие Т гондий инфекции из-за отсутствия (или очень низкое производство) ИЛ-4 на ранних стадиях инфекции, в то время как он усиливает ИФНγ постановка дифференцированных Т-клеток на поздних стадиях инфекции.
В противоположность этому, Робертс и соавт. (1996) сообщили о том, что увеличение смертности происходит в IL-4-/-мышей во время острой стадии инфекции. Тем не менее, на поздней стадии инфекции, большее число кист и более тяжелой патологии в головном мозге были обнаружены в контроле, чем в IL-4-/-мышей. Во время острой стадии инфекции, что было время, что ИЛ-4-/- мыши умерли, увеличение IFN-γ производство наблюдалась в клетках селезенки IL-4-/-мышей по сравнению с теми, у контрольных животных. Таким образом, авторы предположили, что IL-4 играет защитную роль в предотвращении смертности от понижающей регуляции провоспалительных цитокинов производства во время острой стадии инфекции. Причины различных результатов в исследованиях, описанных неясны. Однако, так как генетический фон мышей и штамма T. гондий различаются между этими двумя шпилька-х годов, эти переменные могут способствовать к различным результатам. В поддержку этой возможности, генетический фон мышей был демон-strated, чтобы повлиять на исход Т. гондий инфекции в IL-4-/- Мышей (Александр и др., 1998).
574 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
В дополнении к регуляторным эффектам IL-4 на IFN-γпроизводство, IL-4, недавно было показано, обладает активностью, которая изменяет внутриклеточную репликацию тахизоитов в мышиных макрофагах (Swierczynski и др., 2000) и клеточные линии фибробластов человека (Шавеш и др., 2001). Необходимы дополнительные исследования, чтобы выяснить роль IL-4 в иммунопатогенезе токсоплазмоза.
IL-5
IL-5, усиливает экспрессию IL-2 рецепторов на
B клеток и способствует пролиферации и дифференцировки В-клеток (Суэйн и др, 1988;.. Takatsu и др
1988). IL-5-дефицитной (IL-5-/-) Мыши имели повышенную смертность во время поздней стадии Infec-ции, а их смертность была связана с большим количеством кист и тахизоитов в мозге, по сравнению с зараженным контрольным мышеем (Zhang и Denkers, 1999). IL-12, производство клетками селезенки от мышей, зараженных в ответ на антигены тахизоит в пробирке была заметно ниже в
IL-5-/-чем у контрольных животных, и это снижение коррелирует с селективной утратой В-клеток во время культивирования. Таким образом, ИЛ-5 играет защитную роль против T.gondii, и его защитной роли связана с производством ИЛ-12.
заражённых IL-6-/- мышей, количества мРНК для ИФНγдетектируется RT-PCR были найдены, что signif-icantly меньше по сравнению с контрольными мышами, в то время как количество IL-10 мРНК были выше, чем у контрольных животных (Сузуки и др., 1997). Кроме того, препараты лимфоцитов выделяют из мозга инфицированной IL-6-/- мыши имели значительно более низкие отношения γδ Т-клетки и CD4+ αβ Т-клетки, но более высокие отношения CD8+ αβТ-клетки, чем те, зараженных контрольных мышей (Сузуки и др., 1997). Интересно, что никаких различий не было обнаружено в соотношениях этих Т-клеточных подмножеств в селезенке между этими животными (Сузуки и др., 1997). В другом исследовании, сывороточный ИФНγ уровни были значительно ниже, чем в контроле IL-6-/-мышей на ранней стадии инфекции (Jebbari и др., 1998). Таким образом, защитная активность IL-6 против развития TE, как представляется, из-за его способность стимулировать IFN-γпроизводство (системное в ранней стадии инфекции и в головном мозге в более поздней стадии) и, чтобы вызвать проникновение и накопление различных подмножеств Т-клеток в головном мозге зараженных мышей. По отношению к защитной роли IL-6 против ТЕ, сообщалось, что плодные микроглии человека, обработанные IL-6 ингибирует intracellu-лар репликации тахизоиты в пробирке (Chao и др., 1994), и что IL-6 действует синергически с IFN-γ ингибировать пролиферацию тахизоитов в мышиных астроцитов (Халонен и др., 1998).
IL-6
ИЛ-6 является многофункциональным цитокином, который регулирует различные аспекты иммунного ответа, реакции острой фазы и гемопоэз (Akira и соавт., 1993), и действует в нервной системе (Хирота и др.,
1996). IL-6-мРНК экспрессируется в мозге мышей, зараженных T. гондий (Gazzinelli и др, 1992;. Хантер и др, 1992;. Deckert-Шлютер и др., 1995), и обнаруживается в ликворе этих мышей ( Шлютер и др., 1993). IL-6-дефицитных (IL-6-/-) Мышей формируется значительно большее число T.gondii, кист и областей воспаления, связанные с тахизоитами в мозге, чем сделали контрольный мышей (Сузуки и др, 1997;.. Jebbari и др, 1998). Эти результаты указывают на то, что IL-6 защищает от развития ТЭ, предотвращая образование кист и пролиферацию тахизоитами в мозге зараженных мышей. В головном мозге
IL-10
IL-10 является важным негативным регулятором inflam-matory ответов (Kuhn и др., 1993). IL-10-Defi-Cient (IL-10-/-) мыши погибают в течение острой стадии инфекции с Т. гондий, а их смертность Asso-лем, связанных с развитием тяжелой Immunopathol-логии, опосредованного Th1 иммунных реакций в печени (Gazzinelli и др., 1996) и кишечника (Сузуки и др ., 2000b). Таким образом, IL-10 имеет решающее значение для понижающего регулирования IFN-γопосредованный иммунный ответ и предотвращая развитие патологии, вызванной иммунными реакциями. Когда IL-10-/- Мышей обрабатывал сульфадиазин контроля prolifera Тиона Т. гондий на ранней стадии инфекции, животные пережили острую стадию, но разработали смертельные воспалительные реакции в мозге в
| ИФН-г-ИНДУЦИРОВАННЫЙ эффекторные МЕХАНИЗМЫ | 575 |
поздняя стадия инфекции (Wilson и др., 2005). Таким образом, IL-10 играет важную downregula-Тори роль для предотвращения иммунопатологии в процессе заражения Т. гондий.
Lipoxin (LX) A4
LXA4 является эйкозаноидов продукт, созданный из арахидоновой кислоты. После заражения Т. гондий, мыши дикого типа получают высокие уровни сывороточного LXA4, начиная с начала хронической инфекции. 5-липоксигеназы (5-LO) представляет собой фермент, Crit-ческое в генерации LXA4 и мышей с дефицитом 5-LO (5-LO-/-) Поддались инфекции во время хронической стадии, отмеченные отображения inflamma-ции в мозге (Aliberti и др., 2002). Тем не менее, количество цист в мозге были значительно ниже в 5-LO-/- чем у контрольных мышей, что свидетельствует о том, что смертность в 5-LO-/-мышей не из-за дефектный контроль паразита, но из-за улучшенные воспалительные реакции, вызванных паразитами. Увеличение смертности в 5-LO-/- животных было связано с высотами IL-12 и IFN-γ, И был полностью предотвращено путем введения стабильного аналога LXA4. Поэтому LXA4 имеет важное значение для понижающей регуляции proin-flammatory реакций при хронической стадии Т. гондий инфекции.
МЕХАНИЗМЫ
Несколько механизмов IFN-γиндуцированная активность анти-Toxoplasma в различных клетках-хозяевах, была описана. Эти анти-Toxoplasma эффекторные мехи-ханизмы включают окись азота (NO), производство, trypto-PHAN голодания, генерации активных форм кислорода, железа депривации, и индукции недавно описанных P47 ГТФаз, в том числе IGTP, IRG-47, и LRG-47 , Действия IFN-γ является INITI-ованного путем связывания IFN-γ с IFN-γ рецептор (IFN- &γR) на клеточной поверхности и начало сигнального каскада, с участием семейства JAK из Тайро-синусоидальной киназ и STAT семейства факторов транскрипции. После IFN-γ стимуляция, STAT1 димеризуется и транслоцируется в ядро, где он связывается гамма-активированных элементы последовательности в промоторных областях IFN-γ-inducible генов. Действия IFN-γ опосредованы белков, кодируемых этими
576 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
IFN-γиндуцибельные гены. IFN-γ-inducible гены, которые имеют особое значение для устойчивости к T. гондий, включают в себя гены, участвующие в генерации NO, триптофан деградации и генерации активных промежуточных соединений кислорода, а также гены ENCOD-Инг для P47 ГТФаз, IGTP, LRG-47 и ИРГ-47, как упомянуто выше. Последние данные обнаружили, что STAT1 нулевые мыши (Stat1-/-) Все еще может производить ИФН
γ и разработать некоторые ответы Th1, но STAT1 нулевые мыши не в состоянии активирует многие из antimicro-BIAL эффекторных функций, такие как производство синтетазов оксида азота, индукции некоторых из p47 ГТФаз, и способность контролировать паразит Repli-катион (Гаврилеск и др аль, 2004;.. Либерман и др, 2004). Эти результаты указывают на то, что главная роль STAT1 для развития антимикробного
эффекторные функции, а также выделить важ-стояние из этих IFN-γиндуцированный противомикробный-Mech механизмы надзор в защите против T. гондий.
Исследования показывают, что экспрессия этих антимикробных механизмов различаются в зависимости от ткани, хотя некоторые из этих антимикробных эффекторных механизмов, как были показаны, что важно для контроля T.gondii, в головном мозге. Особое значение для Т. гондия в головном мозге являются гены, кодирующие NO синтеза и IDO-индуцированного триптофан голода, и гены, кодирующие для P47 ГТФаз. Краткое описание каждого из этих механизмов эффек-TOR анти-Toxoplasma и механизма действия каждого приводится ниже.
Продукция NО
НЕТ является одним из основных IFN-γиндуцированная анти-toxoplasmocidal механизмы, как известно, опосредуют устойчивость к T.gondii, у мышей (Gazzinelli и др., 1993). IFN-γиндуцирует синтез фермента, индуцируемого синтетазы оксида азота (INOS), который производит NO из L-аргинина (Adams и др., 1993). L-аргинин-зависимое производство NO, и последующее превращение в нитраты и нитриты, как полагают, имеют прямой antimicrobiocidal активности и привести к гибели паразита. IFN-γиндуцированное NO производство не является одним из основных анти-токсоплазмы эффектора механизма, функционирующий в макрофагах и микроглии, (смотрите раздел 21.7); Однако, мыши, получавшие
с ингибитором иОАС или мыши, дефицитные по иСОА являются относительно более устойчивыми, чем у мышей, дефицитных по IFN-γ, Что свидетельствует о том, что другие эффекторные механизмы, индуцированный IFN-γтакже имеют важное значение (Hayashi и др, 1996;.. Scharton-Керстен и др, 1997). Мыши, дефицитные по NO, однако, более восприимчивы к TE, что свидетельствует о том, что NO играет важную роль в латентной стадии инфекции. НЕТ Также было обнаружено, чтобы вызвать брадизоитную дифференцировку, и, следовательно, NO также может играть определенную роль в поддержании паразита в хронической инфекции (Bohne и др., 1994).
IGTP
В P47 GTPases представляют собой семейство белков, 47-48 кДа в размерах, которые сильно индуцируется IFN-γ(Тэйлор и др., 1996, 2004). Есть шесть членов P47 ГТФ семьи: IGTP, IRG-47, LRG-47, TGTP, IIGP и GTPI. Три из этих P47 ГТФаз, IGTP, IRG-47, и LRG-47, было показано, что важную роль в устойчивости к Т. гондий. IGTP- и LRG-47-дефицитных мышей оба проявили глубокую потерю устойчивости к острой инфекции с Т. гондий, в то время как ИРГ-47 дефицитных мышей отображается только частичную потерю сопротивления, которая не проявлялась до хронической фазы (Кольясо и соавт. , 2001). Кроме того, IGTP и LRG-47 были признаны необходимыми для IFN-γиндуцированная ингибирование Т. гондий в макрофагах, а ИРГ-47 было обнаружено, что нет необходимости (Butcher и др., 2005). Таким образом, эти три p47 GTPases, как представляется, жизненно важные, но различные роли в иммунной защите против T. гондий. Функция IGTP, LRG-47, и IRG-47 не известна, но IGTP локализуется в эндоплазматический ретикулум, предполагая, что IGTP и, возможно, другие P47 GTPases могут быть вовлечены в переработку белка или торговли (Taylor и соавт., 1997). В инфекции Mycoplasma туберкулеза, LRG-47, и было обнаружено, что влияние на подкисление фагосомы, оказывая поддержку идее о том, что P47 GTPases влияют эндоцитических везикулярного торговли людьми внутри клеток-хозяев (MacMicking и др., 2003). Принимая во внимание, что Т. гондий находится в уникальном внутриклеточный компартмент, который обычно не-хозяина с слиянию-клеток везикулярные отсеков, это очень интересная идея, которая заслуживает дальнейшего изучения. IFN-γиндуцированная экспрессия гена IGTP зависит от STAT1, и устойчивость к Т. гондий
| ИФН-г-ИНДУЦИРОВАННЫЙ эффекторные МЕХАНИЗМЫ | 577 |
наблюдалось только тогда, когда IGTP присутствовал в обоих гемопоэтических и негемопоэтических Compart-ке (Кольясо и др., 2002). Это говорит о том, что IGTP необходим в макрофагах и микроглии, а также непрофессиональные эффекторные клетки, как энтеро-cytes, эндотелиальные клетки и астроциты. В отличии от существенной роли IGTP в устойчивости против острой приобретенной инфекции с Т. гондиями, эта ГТФаза была сообщена, чтобы играть только частичную роль в регулировании паразита при хронической стадии инфекции у мышея (Кольясо и др., 2002) , В связи с участием IGTP сопротивления против хронической инфекции, IFN-γ-активированные астроциты были продемонстрированы ингибировать внутриклеточные реплики-Тион тахизоитов в пробирке с помощью механизма зависит-оздействию на IGTP (Халонно и др., 2001).
Утюг лишение
IFN-γбыло показано, ингибирует рост T.gondii, с помощью железа депривации в несколько типов клеток, такие как Ente-rocytes (Dimier и Бут, 1998;. Бут и др, 1999). IFN-γиндуцированная железа лишение было продемонстрировано
578 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
экспериментами, в которых добавление избытка железа через Fe2+ соль была в состоянии преодолеть ИФНγиндуцированная ингибирования, и лечащие клетки-хозяева с хелатор железа десферриоксамина (ДФО) была способна индуцировать ингибирование (Dimier и Бута, 1997). Клетки, как правило, получают железо посредством пути рецептора трансферрина, в котором железа, связанного с трансферрином связывается с рецептором трансферрина на поверхности клеток и комплекса рецептора трансферрина интернализируется с помощью эндоцитоза, с последующим подкислением эндоцитотического пузырька вызывая ионы трехвалентного железа, чтобы отделить от трансферрина. Ионы железа переносятся через мембрану везикулы в цитоплазму, а затем может войти в железный пул клетки. Этот внутриклеточный пул железа доступен для процессов обмена веществ и доступно для комплексообразования, по ДФО. Т. гондий реплицируется в паре-sitophorous вакуоли, мембранный Compart-ние в клетке-хозяине, которая не сливаться с лизосомами и не является смежным с железом трансферриным, как она проходит через эндоцитотический путь. Таким образом, вместо получения железа из трансферрина, эксперименты с энтероцитов показывают, что ИФНγможет ингибировать репликацию Т. гондия путем ограничения-ной доступность внутриклеточного железа для паразита дл выведени из своей клетки-хозяина. -Механич низм, с помощью которого это происходит не понимают. Ингибирование роста Т. гондий через IFN-γиндуцированная железа лишение только было показано, энтеро-cytes, и может быть важным анти-токсоплазматического механизм на поверхности слизистой оболочки в качестве первой линии обороны против патогена вторжения. Не ясно, однако, является ли это IFN-γиндуцированная механизм железа зависит также происходит в других типах клеток и, таким образом имеет более широкую роль в контроле токсоплазмы в латентной стадии инфекции и врожденного токсоплазмоза.
Микроглия
Микроглии являются житель макрофаг населения в ЦНС, и, вероятно, основные эффекторные клетки в предотвращении распространения тахизоит Т. Gondii в головном мозге. Микроглия активируется для ингибирования роста T. гондия в пробирке с IFN-γплюс липополисахарид (Чао и др., 1993a, 1993b, 1994). TNF-αи ИЛ-6, также участвуют в ингибировании T.gondii, в человеческой микроглии (Chao и др., 1994). НЕТ было показано, опосредует ингибирующее действие активированной мышиной микроглии на внутриклеточной репликации тахизоитов, поскольку обрабатывающего единение этих клеток с Nг-monomethyl-L-Argi-девять (который блокирует образование NO) абляции их ингибиторной активностью (Чао и др., 1993а). В последнее время Фрейнда и др. (2001) сообщила об вовлечении-тии как NO-зависимых и -независимых механизмов в сопротивлении мышиной микроглии активированного комбинацией IFN-γ и TNF-α, В отличии от этих наблюдений в мышиной микроглии, было сообщено, что NO не участвует в ингибирующем действии активированного человеческой микроглии против T. гондия (Chao и др., 1994). В естественных условиях, последующие ИНГ Т. гондий инфекция, микроглии активизируются для получения ФНО-αИ IFN-γопосредует Activa-ции (Deckert-SCHLÜTER и др., 1999). IFN-γопосредованный активация микроглии в сотрудничестве с аутокринным TNF-α вероятно, один из механизмов резистентности мозга против T. гондий.
Активированные микроглии производят IL-1β, ИЛ-12 и ИЛ-15, и выражают МНС класса I и II, LFA-1 и ICAM-1 (Шлютер и др., 2001). Таким образом, в дополнение к их функциям в качестве эффекторных клеток, IFN-γ-активированную микроглии может выступать в качестве антиген-представляющих клеток через МНС класса I и II молекулы на их поверхности, а также в качестве функции регуляторных клеток на иммунные ответы через их IL-1β, IL-12 и IL-15 производства.
| Эффекторные клетки в мозге с активностью против T. гондия | 579 |
Астроциты
в пробирке исследования показали, что IFN-γ-активированные астроциты могут ингибировать рост Т. гондий. В человеческих астроцитов, IFN-γ плюс IL-1βстимулировал астроциты, чтобы ингибировать рост T. гондий через NO-опосредованный механизм (Петерсон и др., 1995). TNF-α и IFN-γБыло показано, что для ингибирования роста T. гондий в глиобластомы полученных клеток человека и нативных астроциты с помощью индукции indoleamine 2,3-диоксигеназы (Daubener и др., 1993). Со-активация indoleamine 2,3-dioxyge-Nase (IDO) активности и NO в человеческих астроцитах приводило к ингибированию активности ИДО, что указывает на поперечное регулирование между этими двумя путями в астроциты (Oberdorfer и др., 2003). Мышиные Астро-cytes также было показано, ингибирует рост T. гондий в пробирке (Халонен и др., 1998). В мышиных астроцитов IFN-γ одна индуцировали сущест-косяк ингибирование роста T. гондий, в то время как IFN-γ в сочетании с TNF-αIL-1 или IL-6, действовал синергический, чтобы усилить это торможение. Ни в одном из TNF-αIL-1 или IL-6, в одиночку имел никакого эффекта на рост T.gondii при мышиных астроцитов. Тормозящее действие ИФНγ-активированные астроциты были установлены, не быть опосредованы через NO или IDO (Халонно и Weiss, 2000), как это имело место для человеческих астроцитов или астроцитомов клеток. Механизм IFN-γиндуцированная ингибирование в мышиных астроцитов было также установлено, что независимо от реактивного кислорода интер-опосредует и железа лишения. Механизм IFN-γиндуцированное торможение в мышиных астроцитах было, однако, нашли привлечь ИФНАγиндуцированная p47 ГТФ, IGTP, как IFN-γ Ингибирование было отменено в IGTP-/ -астроциты (Халонен и др., 2001). Как обсуждалось ранее, этот механизм IFN-γиндуцированная-IGTP-зависимость не под стояла в это время, но так как IGTP локализуется в эндоплазматический ретикулум было высказано предположение, что он может быть вовлечен в незаконный оборот молекул в паразитофорной вакуоли. Учитывая тот факт, что астроциты являются доминирующими глиальными клетками в головном мозге, в IGTP-опосредованный бактериостатические эффекты астроцитов могут оказывать значительное влияние на ограничение роста паразита в мозге и позволяя более мощный antimicrobiocidal
деятельность микроглии, чтобы очистить паразитов от мозга.
Эндотелиальные клетки
IFN-γ-стимулированные человеческого мозга микрососудистых эндотелиальные клетки, как было показано, чтобы побудить inhi-bition Т. гондий в зависимости от дозы (Daubener и др., 2001). Этот эффект был противопаразитарное усиливается TNF-α, Хотя TNF-αв одиночку не оказывает никакого влияния на рост паразита. Механизм эффектор в этих клетках, как сообщается, опосредовано через триптофана голодания вследствие индукции IDO. Так как один из первых шагов в развитии церебрального токсоплазмоза является проникновение через гематоэнцефалический барьер, IFN- &γиндуцированная ингибирования в этих клетках, может оказаться важным в ограничении репликации T. гондий в головном мозге.
Дендритные клетки
Т. гондий инфекция приводит к развитию зрелых дендритных клеток, экспрессирующих МНС класса II, CD40, CD54, CD80 и CD86 в мозге мышей. Дендритные клетки, как были показаны, чтобы ингибировать внутриклеточную репликацию тахизоитов через реактивные механизмы кислорода опосредованных в пробирке при активации IFN-γ(Июнь и др, 1993;.. Элин и др, 2002); Таким образом, эти клетки могут функционировать в качестве эффекторных клеток, чтобы контролировать паразит в мозге. Однако, дендритные клетки, вероятно, играют более важную роль в качестве антиген-представляющих клеток и IL-12, произ-ERS. Так как микроглии и астроцитов нужно ИФНγчтобы их экспрессия активирует МНС молекул на их поверхности, и так как GM-CSF (цитокина, что астроциты производят в ответ на Т. гондия; Фишер и Белинская, 1999), индуцирует созревание дендритных клеток в культурах первичных клеток головного мозга с паразитами, дендритными клетки могут играть решающую роль в качестве антиген-представляющих клеток в головном мозге после заражения. С другой стороны, дендритные клетки могут также играть патогенную роль в развитии TE, поскольку недавнее исследование показало, что дендритные клетки отвечают за перевозки отдельного тахи- zoites в мозг (Courret и др., 2006).
580 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
ВЫВОДЫ
IFN-γопосредованный иммунный ответ играет центральную роль в сопротивлении против Т. гондий в головном мозге. IFN-γПроизводство нескольких типов клеток, в том числе Т-клетки, необходимо для сопротивления. Микроглии являются один тип IFN-γ-продуцирующих клетки, которые, вероятно, играют важную роль в борьбе с паразитом. IL-12, Bcl-3, NF-κВ (2), с-Rel и CD40-CD40L взаимодействие лиганда имеют важное значение для повышающей регуляции и от IFN- ОБСЛУЖИВАНИЕγПроизводство Т-клетки для профилактики ТЕ (табл 21.1). С другой стороны, IL-10 и Lipoxin A4, кажется, играют решающую роль в понижающей регуляции ИФНγопосредованная реакция, препятствующие Devel-витие иммунного ответа, опосредованная патология.
IFN-γактивизирует микроглии и астроцитов, чтобы ингибировать внутриклеточный репликацию тахизоитов (рис 21.1). На сегодняшний день три механизма были идентифицированы
| ВЫВОДЫ | 585 | ||
| макрофаг | Т-клетки |
Астроциты или
Тохо
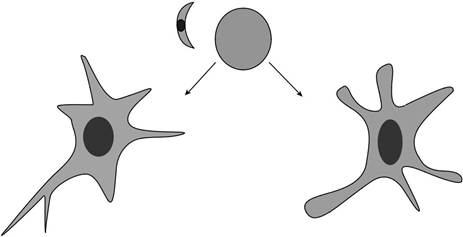
IFN-γ IFN-γ
ФНО-α
ФНО-α
| NO-зависимый | НЕТ-независимый | |
| (Мышь) | ||
| (Мышь) | ||
Рисунок 21.1 Контроль токсоплазма в головном мозге требует два популяций эффекторов.
Таблица 21,1 IFN-γопосредованное сопротивление Т. гондий в головном мозге

| молекула | функция | ячейки | Рекомендации |
| IL-12 | Поддержание IFN-γ Производство Т-клетки | дендритные клетки | Фишер и др., 2000; Яп и др., 2000 |
| Bcl-3 | Поддержание IFN-γ Производство Т-клетки | Т-клетки | Franzoso и др., 1997 |
| NF-κБИ 2) | Поддержание IFN-γ Производство Т-клетки | Т-клетки | Caamaño и др., 2000 |
| с-Rel | повышающая регуляция IFN-γ Производство Т-клетки | Т-клетки, дендритные | Мейсон и др., 2004 |
| ячейки | |||
| CD40L | повышающая регуляция IFN-γ производство по | Т-клетки | Subauste и др., 1999; Райхман |
| Т-клетки (в человеке, но не в мыши) | и другие., 2000 | ||
| IFN-γ | Активация эффекторных клеток для контроля | γδ Т-клетки (?) | Сузуки и др., 1997; Лейва и др., |
| Т. гондий | 1998; Лепаж и др., 1998 | ||
| микроглии и | Сузуки и др., 2005 | ||
| макрофаги | |||
| αβ Т-клетки | Сузуки и др 1989a. Браун и | ||
| Маклеода, 1990; Gazzinelli и др., | |||
| 1992; Кан и Сузуки, 2001; | |||
| Wang и др., 2004, 2005 | |||
| иОАС | Производство NO для управления T.gondii, | Микроглия | Чао и др, 1993a. Фрейнд и др., |
| в эффекторных клеток | (Мышь) | 2001 | |
| астроциты | Петерсон и др., 1995 | ||
| (человек) | |||
| Я ДЕЛАЮ | Триптофан деградация для контроля | астроциты | Daubener и др., 1993 |
| Т. гондий в эффекторных клеток | (человек) | ||
| кровоснабжения головного мозга | Daubener и др., 2001 | ||
| (эндотелиальный | |||
| клетки человека) | |||
| IGTP | Неопознанный механизм контроля | астроциты | Халонен и др, 1998, 2001. |
| Т. гондий в эффекторных клеток | (Мышь) | Халонен и Weiss, 2000 | |

586 Церебральный токсоплазмоз: ПАТОГЕНЕЗ И HOST СОПРОТИВЛЕНИЕ
Контроль Т. гондий в IFN-γ-активированные эффекторные клетки в головном мозге: НЕТ производство на иСОА, триптофан деградации по Идо, и неизвестные механизмы, опосредованных IGTP. Кроме того, астроциты и микроглии играют важную регуляторную роль в IFN-γопосредованного иммунного ответа по производству цитокинов, таких как TNF-α, ИЛ-1, ИЛ-6 и ИЛ-10, и хемокины. Нейроны-видимому, участвуют в этих взаимодействиях. Важно, чтобы выяснить подробные механизмы взаимодействия между Т-клеток, глиальных клеток и нейронов, и молекулярной основы их защитных функций, для того, чтобы получить лучшее представление о immunopathogene-лиза церебрального токсоплазмоза.
БЛАГОДАРНОСТЬ
Мы благодарим Рене Миллер и Брэдли Данфорда за помощь в подготовке рукописи. Эта работа проводится при поддержке службы общественного здравоохранения гранта AI 047730 (YS).
РЕКОМЕНДАЦИИ
Abou-Bacar, А., Pfaff, AW, Letscher-Брю, В. и др. (2004). Роль гамма-интерферона и Т-клеток при врожденной передачи Toxoplasma. Паразит Immunol. 26, 315-318.
Адамс, Л. Б., Хиббс, JB-младший, Тейнтор, РР и Кренбюля, JL (1990). Бактериостатический эффект мышиных активированных макрофагов для токсоплазма. Роль для СЕНТ-лиза неорганических оксидов азота из L-аргинина. J. Immunol. 144, 2725-2729.
Адамс, Л. Б., Fukutomi, Ю. и Кренбюля, JL (1993). Регулирование мышиных макрофагов функций эффекторных по lipoarabinomannan из штаммов микобактерий с различной степенью вирулентности. Infect. Имун. 61, 4173-4181.
Айзенберг Д., Конья, Н., Париж, Л. и др. (2002). Генотип 86 токсоплазма изоляты, связанные с врожденным токсоплазмозом человека, и корреляции с клиническими данными. J. Infect. Дис. 186, 684-689.
Akira, S., Taga, Т. и Kishimoto, Т. (1993). Интерлейкина-6 в биологии и медицине. Adv. Immunol. 54, 1-78.
Александр, J., Jebbari H., Bluethmann, H., Brombacher, Ф. и Робертс, CW (1998). Роль IL-4 у взрослых приобретенного и врожденного токсоплазмоза. Int J. Parasitol. 28, 113-120.
Aliberti J., Reis е Соуза, С., Schito, M. и др. (2000). CCR5, обеспечивает сигнал для микробной индуцированной продукции IL-12 CD8 альфа+дендритные клетки. Природа Immunol. 1, 83-87.
Aliberti, J., Serhan, С. и Sher, A. (2002). Паразит-индуцированной lipoxin А4 является эндогенным регулятором IL-12 производства и иммунопатологии в токсоплазма инфекции. J. Exp. Med. 196, 1253-1262.
Aliberti J., Валенсуэла, JG, Каррузерс, В. Б. и др. (2003). Молекулярная мимикрия в CCR5-связывающий домен в микробной активации дендритных клеток. Природа Immunol. 4, 485-490.
Алин, Ф. Бут, Д. и Dimier-Пуассона, И. (2002). Дендритные клетки в качестве эффекторных клеток: гамма-интерферон активация мышиных дендритных клеток вызывает кислородное-зависимое ингибирование репликации токсоплазма. Infect. Имун. 70, 2368-2374.
Araujo, FG и Slifer, Т. (2003). Различные штаммы токсоплазма вызывают различные реакции цитокинов у мышей линии CBA / Ca. Infect. Имун. 71, 4171-4174.
Биман, MH, Вонг, С. Ю., Абрамс, JS и Ремингтон, JS (1993). Селезеночной секрецию ИЛ-4 не связан с развитием токсоплазматического энцефалита {81} абстрактным. Программа и тезисы 2-го Международного конгресса модификаторов биологического ответа, Сан-Диего.
Bliss, SK, Butcher, BA и Denkers, EY (2000). Быстрое набора нейтрофилов, содержащие предварительно сохранено IL-12 в ходе микробной инфекции. J. Immunol. 165, 4515-4521.
Bliss, СК, Гаврилеск, ЛК, Алькарас, А. и Denkers, Е.Ю. (2001). Нейтрофильная истощение во токсоплазме инфекции приводит к нарушению иммунитета и летальной системной патологии. Infect. Имун. 69, 4898-4905.
Bohne, В., Heesemann, Дж и Гросс, У. (1994). Снижение репликация токсоплазма необходима для индукции брадизоитных специфических антигенов: возможная роль оксида азота в инициировании конверсии стадии. Infect. Имун. 62, 1761-1767.
Бут Д., Moretto, М., Dimier-Пуассона, И. и Gatel, Д. Б. (1999). Взаимодействие между токсоплазмом и энтероцитами. Иммунобиологии 201, 225-228.
Браун, CR и Маклеод, Р. (1990). гены МНС класса I и CD8+Т-клетки определяют количество кист в токсоплазме инфекции. J. Immunol. 145, 3438-3441.
Браун, CR, Дэвид, CS, Кхаре, SJ и Маклеод, R. (1994). Влияние человеческого класса I трансгенов на формирование гондий цист токсоплазм. J. Immunol. 152, 4537-4541.
Браун, CR, Хантер, Калифорния, Эстес, Р. и др. (1995). Окончательное определение гена, который придает резист-ANCE против Toxoplasma кист бремени и encephali-тиса. Immunology 85, 419-428.
Мясник, Б. А., Грин, Род-Айленд, Генри, SC и соавт. (2005). P47 GTPases регулируют токсоплазма выживание в Acti-vated макрофагов. Infect. Имун. 73, 3278-3286.
Caamaño J., Тато, К., Кай, G. и др. (2000). Идентификация роли NF-каппа В2 в регуляции apopto-лизе и в поддержании Т-клеточный иммунитет к токсоплазму. J. Immunol. 165, 5720-5728.
Чакрун, М., Meyohas, МС, Pelosse, B. и др. (1990). Глазной токсоплазмоз СПИД. Энн. Med. Interne (Париж) 141, 472-474.
Чанг, HR и Pechere, JC (1989). Макрофаги oxida-онно метаболизма и внутриклеточного токсоплазма. Микроб. Pathog. 7, 37-44.
Введение
21,2 Производители интерлейкина (IL) -12 требуется для IFN-γ производство
21,3 Производители IFN-γ
Участие других цитокинов и
регуляторные молекулы в сопротивлении 21.5 Вовлечение гуморального иммунитета в
Сопротивление
21,6 IFN-γиндуцированные эффекторные механизмы
Дата: 2019-05-28, просмотров: 369.