Химической связи
Гамильтониан многоэлектронной многоядерной системы в пренебрежении релятивистскими эффектами состоит из членов, описывающих кинетические и потенциальные энергии электронов и ядер. Сложились два подхода к описанию взаимодействий в таких системах – электростатический или силовой (binding) и энергетический (bonding). Силы, действующие в таких системах, подчиняются закону Кулона, причем межэлектронные взаимодействия происходят таким образом, чтобы удовлетворялся принцип Паули. Влияние магнитных сил, обусловленных наличием электронного и ядерного спинов, на полную энергию пренебрежимо мало, не говоря уже о гравитационных силах.
Электростатический подход ищет объяснение химической связи в терминах сил, действующих в молекулах. В энергетическом подходе для этого анализируют изменения кинетической и потенциальной энергий. Оба подхода являются взаимодополняющими и во многом опираются на две следующих важных теоремы.
Теорема вириала устанавливает соотношение между полной энергией многоэлектронной и ее кинетической и потенциальной компонентами. Можно показать, что плотности кинетической g(r) и потенциальной v(r) энергий и электронная плотность r (r) в системе с произвольной ядерной конфигурацией связаны соотношением:
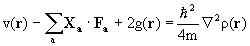 (3.18)
(3.18)
Здесь полная плотность потенциальной энергии v(r)=vNN(R)+ vNe(R, r) +vee(r), Ra –координата ядра а, R=(x, y, z), Fa – сила (вектор), действующая на это ядро. Величина 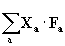 называется вириалом сил, действующих на ядра системы. Если система находится в равновесии, силы, действующие на ядра, равны нулю. Под понятием “вирал” имеют ввиду сумма произведений векторов сил на вектора направлений их действия. Сам вириал – величина скалярная и измеряется в единицах энергии.
называется вириалом сил, действующих на ядра системы. Если система находится в равновесии, силы, действующие на ядра, равны нулю. Под понятием “вирал” имеют ввиду сумма произведений векторов сил на вектора направлений их действия. Сам вириал – величина скалярная и измеряется в единицах энергии.
Интегрируя оставшиеся члены в (3.18) по всему пространству, легко получить следующее соотношение для средних значений компонент полной энергии:
V + 2G =0 или -V = 2G (3.19)
(использована теорема Остроградского-Гаусса, согласно которой объемный интеграл от Ñ 2r (r) по всему пространству равен нулю). Полная энергия системы равна сумме потенциальной и кинетической энергии Е=G+V, отсюда следует соотношение:
Е = - G. (3.20)
Соотношения (3.19) и (3.20) выражают содержание теоремы вириала, характеризует систему в состоянии равновесия. Они справедливы как для полной системы, так и для частей, из которых она образуется; для молекул и атомов. Именно поэтому они полезны при анализе природы химической связи.
Рассмотрим с этих позиций образование молекулы. Для бесконечно удаленных атомов (состояние 1) и для стабильной молекулы (состояние 2) выполняются соотношения:
G1= -(1/2)V1 , G2= -(1/2)V2 . (3.21)
Изменение компонент энергии при образовании молекулы (при абсолютном нуле) равно:
D G = -(1/2) D V. (3.22)
Соответствующее изменение полной энергии
D Е= D G +D V (3.23)
можно связать, используя теорему вириала, с изменением кинетической и потенциальной энергий:
D Е=D G+D V = -(1/2)D V+D V = (1/2)D V,
D Е=D G+D V = D G - 2D G = -D G. (3.24)
Отсюда видно, что изменение полной энергии имеет тот же знак, что и изменение потенциальной энергии. Поскольку при образовании молекулы полная энергия понижается, то это сопровождается понижением потенциальной энергии. Одновременно кинетическая энергия повышается, но ее эквивалентное изменение вдвое меньше по величине (G > 0!). Таков общий баланс изменений энергии, сопровождающих образование молекулы. На этом фундаменте должен строиться дальнейший детальный анализ.
Теорема Гельмана-Фейнмана гласит, что для точной и хартри-фоковской волновой функции справедливо соотношение
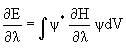 , (3.25)
, (3.25)
где l - некоторый параметр, от которого зависит энергия.
Примение соотношения (3.25) к изучению сил, действующих в молекуле (Гельман, 1936, Фейнман, 1939) известно под названием электростатическая теорема. В качестве параметра используются ядерные координаты: l =Rа. Кинетическая энергия электронов и межэлектронное взаимодействие от координат ядер зависят не явно. Поэтому в этом случае (3.25) дает для ядра а молекулы:
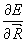 =Za
=Za 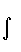 ρ(
ρ( 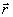 )
) 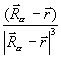 dV +
dV + 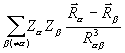 , (3.26)
, (3.26)
где ρ( ) = Ψ*Ψ – электронная плотность.
Производная 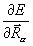 описывает силу, действующую на ядро в молекуле. Ее компонента по одной из осей координат равна
описывает силу, действующую на ядро в молекуле. Ее компонента по одной из осей координат равна
Fxα = – Zα ρ( ) 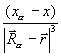 dV +
dV + 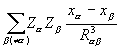 . (3.27)
. (3.27)
Можно заключить, что силу, действующую на ядро а, можно рассматривать классически как сумму электростатического взаимодействия ядра а с электронной плотностью r и с другими ядрами. Поэтому теорему Гельмана-Фейнмана, записанную в форме (3.26), называют также электростатической теоремой.
Из сказанного следует интересный и важный вывод. В классической электростатике показано, что статическая система электрических зарядов не может быть в равновесии (теорема Ирншоу). Тем не менее, если энергия стационарна и получена с помощью вариационного принципа, то ее значение вычисленное как среднее от гамильтониана (как требует квантовая механика) и таковое, вычисленное интегрированием сил, действующих на ядра, по мере их сближения из бесконечности в равновесное положение (т.е. классически), эквивалентны. Это означает, что квантовохимически “правильная” электронная плотность, отвечающая минимуму энергии, в принципе, позволяет получить корректное значение энергии классически с помощью соотношения:

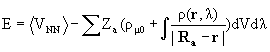 (3.28)
(3.28)
(l 0 характеризует значение, ниже которого система перестает быть стабильной).
В равновесных системах силы, действующие на ядра, равны нулю и анализируя соответствующее электронное распределение, можно выявить некоторые черты химической связи (Берлин, 1951). Рассмотрим применение силового подхода к двухатомным молекулам. Введем величину
f (r) = 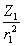 cosθ1 +
cosθ1 + 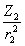 cosθ2 , (3.29)
cosθ2 , (3.29)
являющуюся проекцией на межъядерную ось полной кулоновской силы, действующей в точке г на ядра с зарядами Zi со стороны единичного отрицательного заряда. q - угол между направлением кулоновской силы, действующей на ядро 1 или 2 и межъядерной осью. С помощью этой величины силу F(R), действующую на ядра, разделенные расстоянием R, можно записать через ЭП в виде:
F(R) = 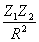 -
- 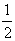 ƒ(r)ρ(r)dV (3.30)
ƒ(r)ρ(r)dV (3.30)
В равновесии R=Re и F(Re)=0. Если принять, что r (г)>0 (отвлекшись от того, что эта функция описывает распределение отрицательного заряда), то знак вклада в значение силы F(R) в каждой точке г определяется знаком проекции полной силы f(r) в этой точке. Отрицательный заряд в областях, где f(r)>0, уменьшает значение F (“связывает” ядра), а в областях, где f(r)<0, увеличивает значение F. Области, в которых f(r)>0 и f(r)<0, называются связывающими и антисвязывающими, соответственно.
В итоге, пространство молекулы можно разделить на связывающую область, в которой ЭП создает электростатические силы, действующие на ядра по направлению друг к другу, и на антисвязывающую область, в которой силы, действующие на ядра, стремятся их раздвинуть. Граничная поверхность, разделяющая связывающую и антисвязывающую области, определяется условием
Fxa=Fxb (3.31)
Для гомоядерной двухатомной молекулы эта поверхность имеет вид двухплоскостного гиперболоида. Области связывания и антисвязывания на плоскости для некоторых молекул показаны на рис.3.15 и 3.16.
Взаимодействие пары атомов сопровождается индуцированием на каждом из них дипольного момента; электронные плотности атомов при этом несколько смещаются друг к другу. Очевидно, что для стабильности системы необходимо, чтобы ЭП в связывающей области при R=Re создавала силы, компенсирующие отталкивание ядер и действие на ядра ЭП в антисвязывающих областях. Исследуем этот вопрос. В большинстве случаев, показанных на рис. 3.8-3.11, образование химической связи сопровождается накоплением ЭП в пространстве между ядрами, которое и отождествляется со связывающей областью: электронный заряд в этом районе стремится приблизить ядра друг к другу. Электроны, находящиеся вне межъядерной области, наоборот стремятся удалить ядра друг от друга, поэтому эти области часто определяют как антисвязывающие. На основании этих аргументов ранее считалось, что требование d r >0 в связывающей области является необходимым (но недостаточным) для образования стабильной системы. Однако имеются примеры таких реально существующих систем, как молекула F2 , для которых характерно наличие минимумов d r в межъядерном пространстве. Можно привести и обратный пример. Вид стандартной карты d r для неустойчивой молекулы Ве2 топографически аналогичен картам для стабильных двухатомных молекул, таких как 02 или N2.
Помимо этого, можно считать твердо установленным, что в некоторых двухъядерных молекулах d r >0 не только в межъядерном пространстве: максимумы d r концентрируются, например, за ядрами. Другими словами, при образовании системы из атомов электроны перетекают и в несвязывающую (с точки зрения силового подхода) область. Такое перераспределение ЭП для двухатомных молекул подтверждается и экспериментальными данными по дифракции электронов на молекулах в газовой фазе.
Таким образом, возникновение стабильной системы обеспечивается весьма тонким балансом сил, действующих на ядра, и соответствующим перераспределением ЭП. Анализ этих сил с позиции перераспределения ЭП, происходящего при образовании химической связи, выявил следующее. Силу, действующую на ядро в двухатомной молекуле, можно записать через ЭП промолекулы r пром и деформационную ЭП d r в виде
F(R) = - ƒ(r)ρпром(r)dV – ƒ(r)δρ(r)dV . (3.32)
Поскольку функция r пром знакопостоянна (положительна с учетом ранее принятого знака для r ) во всем пространстве, а d r — знакопеременная функция, совпадение областей, в которых f и d r имеют одинаковые знаки, должно способствовать стабилизации системы. По этой причине, в частности, области d r <0 в антисвязывающей в силовом смысле области за атомными ядрами на самом деле отвечают связыванию. И, действительно, на картах d r многих молекул имеются минимумы за ядрами на продолжениях линий связи (см., например, рис. 3. 9 - 3.11).
Проанализируем различие в природе связывания в молекулах N2 и F2. В первой из них (рис. 3.8) на карте стандартной d r наблюдается существенная концентрация избыточной ЭП вблизи межъядерной оси и на ее продолжении за ядрами. Во второй молекуле (рис. 3.8) имеются минимумы d r между и позади ядер. Это приводит к различиям в подынтегральных выражениях в последнем члене в выражении (3.27), которые иллюстрирует рис. 3.17. В N2 основной вклад в положительное значение интеграла ò f(r)d r (r)dV, отвечающее связыванию, вносит связывающая область между ядрами, где f(r)>0 и d r (r)>0. Наоборот, в молекуле F2 основное значение этого интеграла определяется областями позади ядер и областями, тороидально охватывающими линию связи вблизи ядер, где f(r) <0 и d r (г)<0. В обеих молекулах важное значение имеют деформации ЭП вблизи атомных ядер, где их характер совершенно различен. Отсюда ясно, что при анализе химической связи нужно рассматривать не только межъядерное пространство, но и распределение ЭП во всех областях исследуемой системы.
Недостатком анализа, основанного на выражении (3.27), является использование не подчиняющихся вариационному принципу функций r пром и d r , поэтому вычисляемые с их помощью компоненты силы могут оказаться физически бессмысленными. В связи с этим иногда целезообразно рассчитывать функции (3.27) для отдельных молекулярных орбиталей: это позволяет соотнести их со связывающими, несвязывающими и антисвязывающими МО. Соответствующие силы имеют в этом случае ясный смысл.
Другой существенный дефект силового подхода в том, что если явно учесть, что ЭП системы r везде отрицательна (а не положительна, как это предполагалось выше), то связывающие и антисвязывающие области следует поменять местами, поскольку заряд ядер остается строго положительным.
Укажем, что силовой подход непригоден для рассмотрения метастабильных состояний, энергия которых выше энергии отдельных атомов: ведь значение силы, действующей на ядро, и в этом случае может быть равным нулю. На вершине энергетического барьера химической реакции, разделяющего исходные и конечные продукты, сила также равна нулю.
Концепция силового связывания не может исчерпывающим образом объяснить возникновение или отсутствие химической связи, поскольку она не позволяет сделать какие-либо выводы относительно изменения кинетической энергии при образовании связи. Перераспределение ЭП, однако, происходит таким образом, что минимизируется полная энергия. Чтобы описать этот процесс, необходимо привлечение иного, энергетического, подхода к анализу природы химической связи.
Первый обстоятельный энергетический анализ, касающийся природы ковалентной связи, принадлежит Рюденбергу, рассматривавшему молекулу Н2 и ион Н2+. Его выводы кратко могут быть суммированы следующим образом. Конструктивная интерференция атомных волновых функций при уменьшении расстояния между атомами приводит к уменьшению кинетической энергии электронов относительно отдельных атомов вследствие уменьшения градиента соответствующих орбиталей. Электронный заряд при этом смещается из областей близ ядер, где потенциал низок, в межъядерное пространство (см. рис. 3.7), увеличивая потенциальную энергию системы. Одновременно ЭП в непосредственной близости от ядер возрастает, в результате чего кинетическая энергия соответствующих электронов увеличивается, а потенциальная - уменьшается. Суммарная энергия системы оказывается ниже, чем у совокупности исходных атомов, причем общий выигрыш в энергии обусловлен понижением кинетической энергии вследствие химического взаимодействия.
Последний вывод, однако, не согласуется с требованием теоремы вириала, согласно которой при образовании стабильной системы из атомов изменение кинетической энергии в среднем положительно, а потенциальной - отрицательно.
Распределение плотностей кинетической и потенциальной энергий электронов в общем случае имеет более сложный характер. Изучая этот вопрос, Бейдер обнаружил, что плотность кинетической энергии при ортогональном преобразовании базиса молекулярных орбиталей ( при переходе к базису естественных орбиталей, в котором матрица плотности диагональна ) имеет вид:
T(r) = - 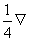 2ρ(r)+G(r). (3.33)
2ρ(r)+G(r). (3.33)
Здесь G(r)= 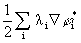 (r)∙
(r)∙ 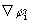 (r)=
(r)= 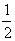 [
[ 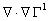 (r, r′)]r=r′=
(r, r′)]r=r′= 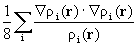 ,
,
ρi(r)=λi 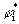 (r)
(r) 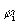 (r), (3.34)
(r), (3.34)
l i - числа заселенности естественных орбиталей. Величина g(r) есть квазиклассическая положительная плотность кинетической энергии, фигурирующая в теореме вириала (3.18). Второй член в (3.33) описывает квантовомеханический вклад в полную плотность кинетической энергии t(r): он имеет разный знак в разных областях пространства. Как мы теперь видим, в теорему вириала (3.18) входят оба эти кинетических члена. Интеграл от (h 2/4m)Ñ 2r (r) по всему пространству равен нулю, поэтому среднее значение кинетической энергии Т равно среднему значению g, обозначаемому как G.
Для анализа компонент энергии связи введем параллельную
T1= 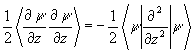 (3.35)
(3.35)
и перпендикулярную
T 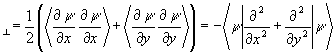 (3.36)
(3.36)
составляющие средней кинетической энергии. Для сферического (изолированного) атома Тô ô = (1/2)Т^ и d =(T^ - Tç ç )/Т = 1/3. Для связанных систем отклонение индекса деформации s от атомного значения 1/3 может служить мерой изменений компонент кинетической энергии, произошедших при образовании химической связи.
Как видно из выражения (3.34), для одноэлектронной молекулы H2+, кинетическая энергия полностью определяется градиентом ЭП Ñ r . Однако уже для двухэлектронной молекулы Н2 разложение по естественным орбиталям дает лишь 98% полной ЭП и её кинетическая энергия только приближенно определяется Ñ r . Из-за накопления ЭП между ядрами в молекуле H2 компонента Ñ r , параллельная линии связи, мала, величина g(r) в этом районе уменьшается по мере продвижения от ядер к центру связи (рис. 3.16); соответственно уменьшается и вклад в Т параллельного линии связи градиента ЭП. Перпендикулярная составляющая градиента ЭП здесь намного больше параллельной из-за сжатия ЭП по направлению к линии связи. За ядрами обе компоненты Ñ r дают большой вклад в g(r), так как ЭП здесь увеличивается примерно одинаково при приближении к ядру. Изменение G(r) по отношению к отдельным атомам между ядрами отрицательно. Такое поведение компонент плотности кинетической энергии отражается и на их средних значениях. В равновесной молекуле H2 Tç ç (Re)<(1/2)T^ (Re), тогда как Tç ç (Re)<(1/2)Tç ç (¥ ) и T^ (Re)<(1/2)T^ (¥ ). Индекс деформации s =0.4740, т.е. в молекуле Н2 он больше атомного значения 1/3. Примечательно, что наименьшее значение Tç ç отвечает в Н2 не равновесному расстоянию (1.4 а.е.), а значению 2.0 а.е. Тем не менее, и при равновесном состоянии Tç ç =0.3043 а.е., т. е. меньше атомного значения 0.3333 а.е. Однако при R<2.0 a.е. начинает заметно возрастать величина T^ . Поэтому в стабильной молекуле Н2 кинетическая энергия будет увеличиваться за счет увеличение G^ , а в силу теоремы вириала это повлечет за собой уменьшение потенциальной энергии. Рюденберг же рассматривал только составляющую кинетической энергии, параллельную линии связи, и его выводы, следовательно, неполны.
Как впоследствии оказалось, анализ, основанный на двух специфических системах - молекулах Н2 и Н2+,-, вообще нельзя перенести на другие, более сложные системы. В этих молекулах в основном состоянии электроны занимают только связывающие МО, не имеющие узлов, в них нет p -связей, неподеленных пар и т.д. Поэтому рассмотрим несколько более общих случаев.
В нестабильной системе Не2 электронами заняты связывающая и разрыхляющая МО и деформационная ЭП в пространстве между ядрами, отражая действие принципа Паули, отрицательна. В этой области как gê ê (r), так и g^ (r), а значит и Tç ç и T^ увеличены по сравнению с атомными значениями, причем Tç ç увеличивается при сближении атомов несколько сильнее, чем T^ . Из-за этого индекс деформации при R=2.0 а.е. Равен d =0.290, т. е. меньше атомного значения 1/3, и плотность кинетической энергии в межъядерном пространстве Не2 систематически выше, чем у отдельных атомов. Таким образом, в Не2 нет, как в Н2, областей пространства, где аккумуляция ЭП пони жает потенциальную энергию и одновременно дает низкую кинетическую энергию.
Молекула N2 дает пример многоэлектронной системы общего вида, где g(r) нельзя и приближенно связать с Ñ r (r), поскольку сумма орбитальных градиентов плотности не сводится к градиенту их суммы. Распределение g(r) в межъядерном пространстве определяется как связывающими, так и разрыхляющими МО, занятыми электронами (рис. 3.19). Наличие узловых поверхностей у разрыхляющих s -МО в центре межъядерного расстояния приводит здесь к заметному вкладу их градиента в компоненту плотности кинетической энергии, параллельную линию связи. p -МО также дает вклад в gê ê (r). Таким образом, размягчение Ñ r в таких системах еще не означает малости компоненты плотности кинетической энергии gê ê (r).
Профиль разности d g(r)= g(r)мол -å gатом(r) показывает, что d g в центре межъядерного пространства, где в N2 аккумулируются электроны, увеличивается по сравнению с отдельными атомами. В окрестностях ядер картина распределения d g сложнее: плотность кинетической энергии оказывается отрицательной как в области уменьшения ЭП, со стороны связывающей (в силовом подходе) области, так и в областях концентрации ЭП позади ядер, т. е. в антисвязывающих областях, отождествляемых с положением неподеленных электронных пар атомов N. Таким образом, концентрация электронов вдоль оси двухатомной молекулы в антисвязывающих областях понижает кинетическую энергию относительно отдельных атомов. Интересно, что такое сложное распределение плотности кинетической энергии приводит в итоге к тому, что в молекуле N2 индекс деформации d =0.3331, т. е. он практически не отличается от атомного значения.
Таким образом, кинетическая энергия в многоэлектронных системах распределена совершенно иначе, нежели в молекуле Н2. Размягчение градиента ЭП вдоль линии связи не является существенным фактором при образовании связи. Следовательно, молекула Н2 не может служить образцом при изучении природы химической связи. Более того, как видно из приведенных выше выражений, плотность кинетической энергии g(r) зависит не только от градиентов естественных орбиталей, но и от лапласиана ЭП. В некоторой точке пространства знаки Ñ 2r (r) и g(r) могут как совпадать, так и отличаться, причем знак g(r) зависит от знака произведения Ñ j ∙Ñ j *. Для образования химической связи необходима благоприятная контраградиентность орбиталей, описывающих связь.
Для анализа причин возникновения химической связи необходимо рассматривать не только центральные области вдоль межъядерных векторов, но и ЭП вблизи ядер. ЭП именно в этих областях в основном определяет величины сил, действующих на ядра, а общая картина d r отражает изменения полной ЭП, приводящие к обсуждавшимся выше изменениям энергии. Рюденберг высказал мнение, что вблизи ядер происходит сжатие ЭП, увеличивающее кинетическую энергию электронов в этой области. Однако позже было установлено, что такое сжатие характерно лишь для молекулы Н2. В общем же случае, в молекулах, состоящих из легких атомов от Н до F, s -плотность атомных остовов поляризуется в направлении, противоположном дипольной деформации валентных электронных оболочек. В таких молекулах результирующее действие электростатических сил, порождаемых p -МО, почти равно нулю. В согласии с этим выводом именно такое распределение ЭП найдено у s -МО молекулы N2 (рис. 3.20).
Силы, действующие на ядра, сильно зависят от числа валентных электронов во взаимодействующих атомах и соответственно от характеристик волновых функций, описывающих эти электроны: главного, орбитального и магнитного квантовых чисел. Именно эти характеристики во многом определяют особенности распределения d r , которые обычно и пытаются соотнести с характером химической связи. При взаимодействии атомов, имеющих 3s- и Зр-электроны во внешней оболочке, конструктивная интерференция в области s -связи будет более слабой, чем атомов второго периода таблицы Менделеева. Причина этого кроется в большом числе узлов у радиальных волновых функций у первых из них. В то же время, p -взаимодействие атомов в обоих случаях не должно существенно отличаться. Причем, если в системе есть занятые электронами p -орбитали, то они, в основном обеспечивают связывание, поскольку влияние s -орбиталей в значительной степени взаимно компенсируется.
В таблице 3.5 приведены значения компонент gê ê (rс) и g^ (rс) в критических точках (3, -1) в молекулах с разными типами химической связи. Отношение этих величин отражает отношение компонент кривизны ЭП в критической точке связи. Оказалось, что для атомных взаимодействий ковалентного типа gê ê (rс) < g^ (rс), тогда как для взаимодействий ионного типа gê ê (rс) > g^ (rс). Кроме того, плотность кинетической энергий, приходящаяся в критической точке (3, - 1) на единицу электронного заряда, g(rс)/ r (rс)>1 для взаимодействий ионного типа и g(rс)/ r (rс)<1 для взаимодействий ковалентного типа. Это означает, что если положительная кривизна ЭП велика в результате сжатия ЭП к ядрам, то величина g(rс)/ r (rс) также достаточна велика, причем величина ее компоненты, параллельной линии связи, превышает величину перпендикулярной компоненты. Для ковалентных взаимодействий наблюдается обратная картина и отношение g(rс)/ r (rс) мало. Значение g(rс)/ r (rс) для молекулы H2 аномально мало: эта молекула нетипична и в этом случае.
Интересное поведение демонстрирует плотность кинетической энергии молекулы F2. В критической точке связи Ñ 2r (rс)= 0.233, l 1=l 2=-0.731, l 3=1.694 а.е. Отношение g(rс)/ r (rс) равно 0.86, как и в молекуле N2, однако g^ (rс)/gê ê (rс)= 0.751, что значительно меньше, чем в случае атомных взаимодействий ковалентного типа.
Резюмируя, следует подчеркнуть, что электростатические эффекты можно считать первопричиной образования химической связи из-за связывающих сил, действующих на ядра в промолекуле. Однако конкретный тип образующейся химической связи, определяемый сортом и числом взаимодействующих атомов, формируется под контролем принципа Паули в результате квантовомеханической интерференции электронных атомных облаков, которая может носить как конструктивный, так и деструктивный характер. Результирующее распределение ЭП отражает совокупность этих факторов и определяет энергетические особенности образовавшейся химической связи. Именно поэтому характеристики ЭП используются для идентификации химических связей.
Многоатомные молекулы
Молекулы, образованные из нескольких атомов, характеризуются, в общем случае, нелинейной и неплоской ядерной конфигурацией. МО этих молекул представляют собой делокализованные одноэлектронные функции, построенные из АО разных атомов (канонические МО). Эти функции не позволяют объяснить строение молекул, трансферабельность и аддитивность свойств функциональных групп. Чтобы проанализировать структуру и химическую связь в таких молекулах, существует ряд полуколичественных или качественных понятий. Рассмотрим основные из них.
Дата: 2019-02-19, просмотров: 409.