Параметризация методов CNDO и INDO не позволяет воспроизводить с их помощью теплоты образования, орбитальные энергии и спектры. Поэтому эти методы не пригодны для построения поверхностей потенциальной энергии, т.е. для исследования относительной устойчивости молекул и механизмов реакций. Дьюар с сотр. (1975) модифицировали метод INDO, изменив параметризацию интегралов b m n и g AB таким образом, чтобы обеспечить возможность расчета перечисленных свойств. Так, резонансные интегралы b m n рассчитываются в MINDO по формуле b m n =GAB (Im +Im )Sm n , где GAB – безразмерный эмпирический параметр, характеризующий типы взаимодействующих атомов. Двухэлектронные кулоновские интегралы g AB вычисляются по модифицированной формуле (2.68). Кроме того, отталкивание атомных ядер вычисляется с учетом их экранирования электронами остовов.
Матричные элементы оператора Фока в методе MINDO для систем с закрытыми оболочками в пренебрежении интегралами проникновения приведены в таблице 2.16.
Существуют различные параметризации метода MINDO, из которых наиболее известной является схема MINDO/3. Параметризуемыми свойствами здесь служат теплоты образования, причем параметры зависят от свойств как атомов, так и их парных комбинаций. Кроме того, орбитальные экпоненциальные множители, используемые для расчета интегралов, также являются параметрами. В итоге, теплота образования воспроизводится в методе MINDO/3 с “химической” точностью ~ 4 ккал/моль а потенциалы ионизации - ~ 0.35 эВ. Геометрия молекул также предсказывается довольно точно (табл. 2.17). В тоже время, спектральные характеристики, водородные связи и описание отталкивания неподеленных электронных пар остаются слабым местом MINDO/3.
Таблица 2.17. Теплоты атомизации и геометрические характеристики некоторых молекул
Молекула
ΔΗ
Геометрия. Å и градусы
Расчетная
Экспериментальная
Углы и связи
Расчетная
Экспериментальная
Метод Модифицированного Пренебрежения Дифференциальным
Перекрыванием (MNDO).
Чтобы более корректно учесть отталкивание неподеленных электронных пар, Дьюар с сотр. (1977) предложил включить в расчет все двухэлектронные интегралы, содержащие пары АО, принадлежащие одному и тому же атому; перекрывание АО различных атомов по-прежнему игнорирутся (приближение двухатомным дифференциальным перекрыванием). Модификация коснулась далее способа выбора параметров: в отличие от MINDO/3, они зависят только от свойств отдельных атомов, а не от их парных комбинаций. Это позволяет параметризовать метод по большему числу соединений, расширяя таким образом сферу его применимости.
В результате развития этого метода появились различные схемы, отли-чающиеся выбором параметров: AM1, AM3 (Austin Model), PM3 (Parameterised Model 3) и др. Они обеспечили возможность расчета водородной связи (AM3), а также энергии образования органических молекул и переходных состояний органических реакций (PM3) с ошибкой менее 5 ккал/моль.
2.21 Разделение s - и p -электронов. p -электроннное приближение
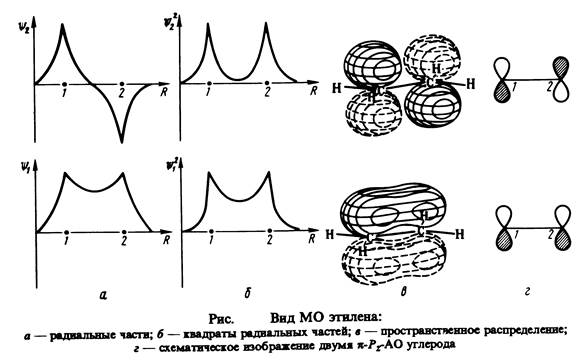 При квантовохимических исследованиях ненасыщенных и ароматических молекул, чаще всего являющихся плоскими, как правило, используют s , p - приближение, состоящее в следующем. Для плоских молекул все валентные АО можно разбить на две группы. Одна из них содержит орбитали, симметричные относительно отражения в плоскости молекулы ( s -АО ), другая - орбитали, антисимметричные относительно такого отражения ( p -АО). Рис.2.7 иллюстрирует это на примере этилена. s -электроны имеют максимальную вероятность нахождения в плоскости молекулы и поэтому локализованы близ ее, вероятность нахождения здесь p -электронов, наоборот, равна нулю. p -электроны слабее связаны с остовом молекулы, более подвижны, легче ионизируются и более активны во взаимодействиях.
При квантовохимических исследованиях ненасыщенных и ароматических молекул, чаще всего являющихся плоскими, как правило, используют s , p - приближение, состоящее в следующем. Для плоских молекул все валентные АО можно разбить на две группы. Одна из них содержит орбитали, симметричные относительно отражения в плоскости молекулы ( s -АО ), другая - орбитали, антисимметричные относительно такого отражения ( p -АО). Рис.2.7 иллюстрирует это на примере этилена. s -электроны имеют максимальную вероятность нахождения в плоскости молекулы и поэтому локализованы близ ее, вероятность нахождения здесь p -электронов, наоборот, равна нулю. p -электроны слабее связаны с остовом молекулы, более подвижны, легче ионизируются и более активны во взаимодействиях.
Свойства ненасыщенных и ароматических систем – высокая реакционная способность, зависимость от донорных и акцепторных заместителей, спектры и т.д – определяются, в основном, именно электронами, описываемыми p -орбиталями. Поэтому при решении уравнений Рутана для таких систем вводят p -электронное приближение (Хюккель, 1931): s -АО считают неполяризованными и включают в атомный остов, а движение p -электронов рассматривают в потенциальном поле таких остовов. Волновая функция молекулы при этом представляется как произведение y = y s y p , где y s и y p - нормированные антисимметричные по отношению к s - и p -электронам функции, соответственно. Их можно разложить по слейтеровским детерминантам, составленных только из s - и только из p -МО. Волновая функция y s одинакова как для основного, так и для возбужденных состояний и все изменения связываются с p -электронами. Существенно, что рассмотрение только p -электронов удовлетворяет вариационному принципу (Мак-Вини, 1954; Лайкос и Парр, 1956).
В результате размерность уравнений Рутана сильно сокращается: например, для этилена вместо 12 валентных электронов необходимо учитывать только 2 p -электрона.
Метод Парризера-Попла-Парра
Объединение нулевого дифференциального перекрывания и p -электронного приближения приводит к методу Парризера-Попла-Парра, дающего прекрасные результаты для p -электронных систем. Матричные элементы оператора Фока для этого метода приведены в табл. 2.16. Um m равно потенциалу ионизации атома в соответствующем валентном состоянии, взятому с обратным знаком: - Im . Одноцентровые кулоновские интегралы g m m оцениваются по формуле Паризера-Парра (2.66). Величины Im и Am , необходимые для расчета Um m и g m m определяют из спектроскопических данных для валентных состояний атомов, используя модель локализованных связей. Двухцентровые интегралы g AB рассчитывают по формулам (2.67)-(2.68). Величины hm n считаются параметрами и выбираются по разному для расчета свойств основного (метод Попла) и возбужденных (метод Парризера-Парра) состояний.
1. Для основного состояния hm n являются резонансными интегралами b m n = kSm n , где k подбирается так, чтобы наилучшим образом воспроизводить теплоты образования в представительном круге соединений (существует и ряд других параметризаций).
2) Для возбужденных состояний следует учесть, что волновая функция молекулы должна быть суммой волновых функций основного Y 0 и возбужденных Y i® k состояний: Y =Y 0 + å аi® kY i® k. Обычно учитывают несколько однократно возбужденных электронных конфигураций заданной мультиплетности и ищут коэффициенты аi® k вариационным методом. Матричные элементы (Y 0ï h p ï Y 0) дают энергию основного состояния Е0, а элементы (Y i® kï h p ï Y j® l) имеют вид:
(Y i® kï h p ï Y j® l)º hp i® k, j® l = d kle k - d ije i +2(jkï li)-(jk ï il). (2.70)
В приближении НДП
hp i® k, j® l = d ij d kl (e k - e i) +å [2(1-R)сm j сm l cn i сn k - сm j сm i сn k cn l], (2.71)
где R=0 для синглетных состояний и R=1 – для триплетных.
 Расчет проводится методом ССП, причем вначале определяют МО для основного состояния, а затем, используя их, строят волновые функции возбужденных состояний.
Расчет проводится методом ССП, причем вначале определяют МО для основного состояния, а затем, используя их, строят волновые функции возбужденных состояний.
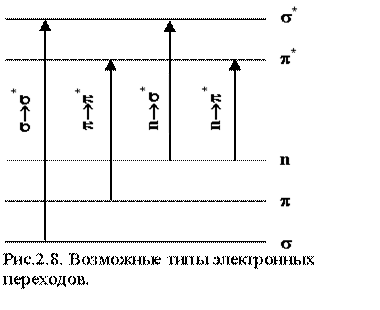
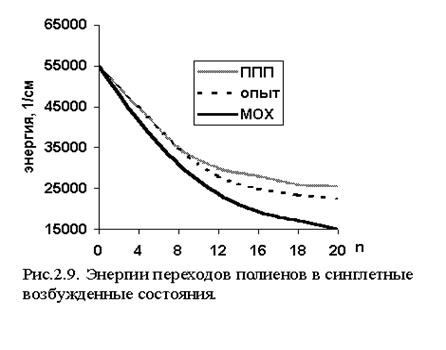
Метод Парризера-Попла-Парра включает в себя метод КВ и очень хорошо зарекомендовал себя как при определении геометрии, потенциалов ионизации и сродства к электрону, так и при расчетах оптических спектров поглощения сопряженных органических молекул. Спектр поглощения состоит из нескольких полос, связанных с определенными электронными переходами. Для плоских молекул МО можно разделить на три группы: s , p и n* . Наболее вероятное относительное расположение соответствующих энергетических уровней и разрешенные правилами отбора типы электронных переходов показаны на рис. 2.8. Диагонализизация матрицы hp i® k, j® l называемой матрицей конфигурационного взаимодействия, дает энергии спектральных переходов и веса возбужденных конфигураций аi® k. Ориентированный на p -электроны, метод ППП хорошо описывает p -p * переходы (рис. 2.9.): точность оценки синглет-синглетных переходов составляет 0,1-0,2 эВ или 3-5% .
 Интенсивность полосы поглощения f определяется квадратом дипольного момента перехода и равна f =Кn ï å å аi® k cn i сn k xï 2. Она оценивается методом ППП с ошибкой 40-50%.
Интенсивность полосы поглощения f определяется квадратом дипольного момента перехода и равна f =Кn ï å å аi® k cn i сn k xï 2. Она оценивается методом ППП с ошибкой 40-50%.
Метод МО Хюккеля
Этот чрезвычайно простой не-ССП метод, первоначально предложенный для углеводородов (Хюккель, 1931), основан на нескольких очень сильных приближениях:
1) Принимается p -электронное приближение; считают, что АО образуют ортонормальный базис, т.е. Sm n = d m n .
2) Межэлектронными взаимодействиями (т.е. всеми двухэлектронными кулоновскими и обменными интегралами) пренебрегают. Из-за этого решение уравнений метода не требует итераций и проводится в один шаг.
1. Матричные элементы оператора Фока оцениваются на основании эмпирической информации и являются фиксированными:
hm m = a m ,
hm n = kb m n . (2. 72)
a m называется кулоновским интегралом (его не следует путать с двухэлектронными кулоновскими интегралами g AB), он принимается равным потенциалу ионизации электрона на орбитали m в свободном атоме.
4) Принимается, что b m n =0, если АО m и n не принадлежат ковалентно связанным атомам.
Соответствующие этим приближениям уравнения Рутана имеют вид
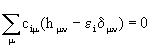 (2.73)
(2.73)
и называются уравнениями Хюккеля, имеющими ненулевые решения при равенстве нулю детерминанта
ï hm n - e id m n ï = 0. (2.74)
Полная энергия в этом методе есть просто сумма орбитальных энергий
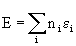 , n=0, 1 или 2, (2.75)
, n=0, 1 или 2, (2.75)
где n – число электронов на МО.
Уравнения Хюккеля могут быть легко записаны и решены для любой системы. Рассмотрим пример молекулы этилена С2Н4, имеющей 2 p -электрона (свяжем их с c (рz) -АО атомов углерода, направленными перпендикулярно плоскости молекулы). Интегралы a С = -11,0 эВ, b СС –2,4 эВ. Детерминант (2.74) имеет вид
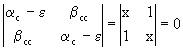 (2.76)
(2.76)
(здесь введены обозначения х=(a С - e )/b СС). Раскрывая определитель, имеем:
х2 –1=0 и х = ± 1, т.е. e 1= a С +b СС , e 2 =a С - b СС.
В принятых обозначениях система уравнений (2.73) имеет вид
с1 х + с2 = 0
с1 + с2 х = 0, (2.77)
Подставим теперь х = ± 1 в (2.77). При х= -1 имеем с1 = с2 . Используя условие нормировки волновой функции этилена с12 + с22 = 1, получаем с1 = с2 = 1/Ö 2 . Таким образом, одна из p -МО этилена имеет вид
j 1 = 1/Ö 2 (c 1 +c 2). (2.78)
При х = 1 имеем с1 = - с2 и, повторяя рассуждения, получаем другую p -МО.
j 2 = 1/Ö 2 (c 1 - c 2). (2.79)
Так как b СС < 0, то e 1 < e 2 причем, e 1 - e 2 = 2b СС. Это означает, что МО j 1 более энергетически стабильна.
Полинг, Уэлланд, Стрейтвизер, Дьюар и другие предложили различные модификации метода Хюккеля, распространив его, в частности, на системы с гетероатомами в цикле. Модификация, в основном, касалась способа выбора параметров a и b и подробно описана в литературе (М. Дьюар. Метод молекулярных орбиталей в органической химии).
Метод Хюккеля, безусловно, является лишь качественным: он ограничен предсказанием энергетики МО сопряженных систем и не способен дать информацию о молекулярной структуре. Однако за счет удачной параметризации этот метод может давать хорошие относительные орбитальные энергии для рядов p -электронных органических и металлоорганических систем. Это позволяет, в частности, идентифицировать полосы в электронных спектрах поглощения таких молекул, отождествляя разности орбитальных энергий e k - e i с энергиями переходов.
Расширенный метод Хюккеля
Наиболее радикальная модификация метода Хюккеля принадлежит Р. Хоффману (1963). Он предложил сохранить оригинальную схему Хюккеля, но включить в рассмотрение все валентные (а не только p ) орбитали и явно учесть интегралы перекрывания. В итоге уравнения метода, получившего название расширенный метод Хюккеля (РМХ), формально совпадают с уравнениями Рутана (2.13), однако содержание матричных элементов совершенно иное. Матричные элементы оператора Фока Fm n º hm n являются параметрами или оцениваются с помощью соотношений, включающих эти параметры. Наиболее часто используются следующие оценки:
hm m = - Im . ,
hm n = 0.5K(hm m + hn n ). (2.80)
В варианте Вольфсберга-Гельмгольца К=1,75. Интегралы перекрывания вычисляются аналитически со ОСТ.
Энергия молекулы с закрытой оболочкой в методе РМХ является удвоенной суммой энергий занятых МО и описывается выражением
Е= 2( å å с2im hm m + å å å сim сin hm n ), (2.81)
в котором члены, ответственные за межэлектронное и межъядерное взаимодействие, не учитываются. В системах с равномерным распределением электронов выражение (2.81) дает хорошие относительные оценки энергии в рядах соединений. Гетероатомы нарушают равномерность электронного распределения и РМХ здесь часто непригоден. Тем не менее расширенный метод Хюккеля дает зачастую лучшие результаты, чем его простой аналог, при изучении конформаций циклических молекул, барьеров внутреннего вращения, относительный порядок уровней энергии.
Уместно отметить роль, которую РМХ сыграл в развитии химии. Именно с его помощью было установлен механизм связывания в металлоценах, сфомулировано правило Вудворда-Хоффмана, исследованы свойства основных состояний многих алифатических и ароматических систем и др.
Программа, реализующая расширенный метод Хюккеля имеется в Интернет: gopher: //infomeister.osc.edu:73/11/software/SOUR CES/FORTRAN/EHT
Дата: 2019-02-19, просмотров: 572.