Органическая химия
Часть 1
УГЛЕВОДОРОДЫ.
ГАЛОГЕНПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Рекомендовано
научно-методическим советом
университета в качестве учебного пособия
Ярославль
2012
ББК 24.23
О 64
УДК 547
Рецензенты: кафедра “Бионеорганическая и биофизическая химия” Ярославской государственной медицинской академии; В.Н.Казин, канд. хим. наук, доцент кафедры общей и биоорганической химии Ярославского государственного университета.
А.Ф.Бетнев, И.С. Колпащикова, Е.Р. Кофанов, Е.М.Алов
О 64 Органическая химия. Часть 1. Углеводороды. Галогенпроизводные углеводородов: Учебное пособие / Яросл. гос. техн. ун-т.- Ярославль, 2012.- 182 с.
ISBN 5-230-17813-2
В пособии рассмотрены основные вопросы строения и реакционной способности, типичные реакции и основные способы получения ациклических, карбоциклических и ароматических соединений, а также галогенпроизводных углеводородов. Пособие составлено с учетом важнейших достижений органической химии за последние годы.
Предназначено для студентов специальности «Химическая технология органических веществ», а также может быть рекомендовано для студентов заочной и дневной формы обучения всех специальностей химико-технологического факультета.
Ил. 18. Табл. 17. Библиогр. 5.
О 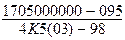 Без объявл. ББК 24.23
Без объявл. ББК 24.23
ISBN 5-230-17813-2 ã Ярославский государственный
технический университет, 2012
Введение
Значение химии вообще, и органической химии в особенности, трудно переоценить. Еще в 18 веке великий русский ученый М. В. Ломоносов говорил: «Широко простирает химия руки свои в дела человеческие» и с этим нельзя не согласиться – нет такой отрасли человеческой деятельности, которой бы не коснулись достижения химии.
Органическая химия – это химия соединений углерода. Вначале органическая химия определялась как химия соединений, которые образуются живой материей. Вплоть до середины 19 века многие химики считали, что органические соединения могут образовываться только в живых организмах и, следовательно, их нельзя синтезировать из неорганических веществ.
В настоящее время большинство органических соединений получают синтетически, используя в качестве сырья, в основном более простые органические соединения. Природными источниками простейших органических соединений являются нефть и уголь.
Что же характерно для соединений углерода и заставляет рассматривать их отдельно от соединений остальных ста с лишним элементов периодической системы? Прежде всего, число органических соединений во много раз превышает число неорганических соединений. А молекулы органических соединений могут быть очень большими по размеру и сложными по строению.
Каковы же особенности углерода, позволяющие ему образовывать столь большое число соединений? Атомы углерода могут соединяться друг с другом, как не могут соединяться атомы никакого другого элемента. Атомы углерода могут образовывать цепи из тысяч атомов или кольца любого размера; цепи и кольца могут иметь разветвления и перекрестные связи. Углеродные атомы, участвующие в образовании этих цепей и колец, могут быть связаны с другими атомами, в основном с водородом, а также с кислородом, азотом, серой, фосфором, галогенами и другими элементами.
Классификация органических реакций
Органические реакции классифицируют по различным признакам:
- по типу превращения субстрата;
- по типу активирования;
- по характеру разрыва связей.
Классификация по типу превращения субстрата
Реакции замещения
Замещение – реакция, в ходе которой атом водорода (или функциональная группа) в органической молекуле замещается на какую-либо функциональную группу (или атом водорода).
Реакции замещения обозначают латинской буквой S (от англ. «substitution» - замещение.
Реакции присоединения
Присоединение – реакция, в ходе которой реагент присоединяется по кратной связи (С=С, С=О, С=N) молекулы субстрата.
Реакции присоединения обозначают латинским символом Ad (от англ. «addition» - присоединение.
Реакции элиминирования
Элиминирование – реакция, в ходе которой от субстрата отщепляется молекула или частица (вода, галогеноводород и т.д.) Этот тип превращения обозначают латинской буквой Е (от англ. «elimination» - элиминирование, отщепление.
Алканы
Алканами называются углеводороды с открытой цепью, имеющие общую формулу CnH2n+2 и содержащие только простые углерод-углеродные связи. Алканы образуют гомологический ряд, в котором каждый член отличается от предыдущего на постоянную структурную единицу -(CH2)- , называемую гомологической разностью.
Таблица 5.1
Физические свойства
Физические свойства алканов определяются их строением. Ковалентные связи в алканах неполярные (С-С) или малополярные (С-Н). Эти связи симметричны, полярность их взаимно компенсируется, поэтому молекула алкана неполярна. Силы межмолекулярного притяжения сравнительно слабы. Это является причиной небольших значений плотности, температуры кипения и плавления.
Температуры кипения, плавления и плотность увеличиваются с увеличением молекулярной массы. Алканы С1 ...С4 - газы, С5 ...С16 - жидкости, далее - твёрдые кристаллические вещества. За исключением низших алканов, температура кипения повышается на 20 - 30 ºС с ростом углеродной цепи на один атом углерода. Такая закономерность изменения температуры кипения характерна для гомологических рядов других классов соединений.
Изомер с разветвлённой цепью имеет более низкую температуру кипения, чем изомер нормального строения.
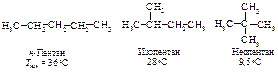
Понижение температуры кипения у изомеров с появлением разветвлений и увеличением их числа объясняется тем, что с увеличением разветвлений форма молекулы стремится к сферической, то есть уменьшается её поверхность, а следовательно, и межмолекулярные силы притяжения. Такая зависимость между формой молекулы и температурой кипения наблюдается и для других классов соединений.
Алканы, неполярные соединения, растворимы в неполярных растворителях (бензол, тетрахлорметан), но нерастворимы в полярных растворителях.
Химические свойства
Химические свойства алканов определяются их строением. В молекулах алканов ковалентные связи малополярные и слабополяризуемые. Поэтому они нечувствительны к ионным реагентам, инертны по отношению к кислотам, основаниям, окислителям. Для взаимодействия с ионными реагентами требуется достаточно высокая полярность атакуемых химических связей, обеспечивающая электростатическое притяжение разноименно заряженных активных центров, и/или способность атакуемой связи поляризоваться под влиянием заряда ионного реагента. Алканы в ничтожной степени обладают и тем и другим свойством.
Наиболее характерным свойством алканов является радикальное замещение незаряженного атома водорода при действии незаряженных радикальных реагентов: атомов хлора и брома при галогенировании, NO2· при нитровании и т.д.
5.2.1. Галогенирование
Замещение водородных атомов на галогены – одна из наиболее характерных реакций предельных углеводородов. Алканы реагируют со всеми галогенами. Со свободным фтором реакция идет со взрывом. Возможны взрывы и в реакциях с хлором. В случае иода процесс ограничен равновесием, так как иодистый водород восстанавливает образующиеся иодистые алкилы.
Галогенирование метана
Хлорирование метана происходит при освещении ультрафиолетовым светом или при повышенной температуре 250 - 400 оС.
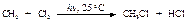
Механизм реакции:
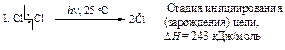
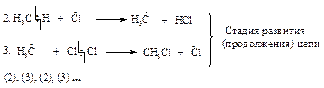
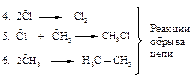
На первой стадии молекула хлора распадается на два атома. Альтернативная возможность разрыва молекулы хлора с образованием ионов хлора (гетеролитический разрыв) не может осуществиться, поскольку для этого требуется значительно большая энергия.
Каждый атом хлора, образующийся в результате гомолитического разрыва связи, сохраняет один электрон из пары, за счет которой осуществлялась ковалентная связь.
Aтом или грyппaатомов, имeющaя нecпaренный электрон, называется свободным радикалом. Неспаренный электрон обозначают точкой.
Атом хлора очень реакционноспособен, так как стремится получить электрон для завершения электронной оболочки. При столкновении с молекулой метана он вырывает атом водорода с его электроном, образуется новый радикал СН3· (стадия 2), который также стремится завершить электронную оболочку и реагирует с молекулой хлора, что приводит к образованию хлористого метила и атома хлора (стадия 3). Реакции 2 и 3 повторяются.
При обрыве цепи реакционная способность реагирующих частиц утрачивается ввиду рекомбинации (объединения) атомов и свободных радикалов в валентно насыщенные молекулы (реакции 4, 5, 6). Поэтому для поддержания реакции требуется постоянное инициирование. В результате рекомбинации двух свободных метильных радикалов образуется побочный продукт - этан. Содержание этана в реакционной смеси невелико, так как стационарная концентрация метильных радикалов, создаваемая в условиях реакции, ничтожна мала.
При хлорировании метана одна реакция инициирования вызывает последовательность реакций, в каждой из которых регенерируется реакционноспособная частица - радикал, вызывающий следующую стадию. Такой механизм называется радикально-цепным. В благоприятных условиях хлорирование метана может пройти от 100 до 10000 циклов прежде, чем произойдет обрыв цепи.
Скорость цепной реакции сильно снижается в присутствии соединений, которые могут взаимодействовать с радикалами и превращать их в мало реакционноспособные частицы. Такие вещества называют ингибиторами. Например, кислород действует как ингибитор.

Радикал СН3ОО· значительно менее реакционноспособен, чем радикал СН3·, и не может продолжать цепь.
Нитрование
Нитрование алканов проводится при повышенной температуре с использованием в качестве нитрующего агента разбавленной азотной кислоты или окислов азота.
Реакция протекает по свободнорадикальному цепному механизму. При этом образуется смесь продуктов.
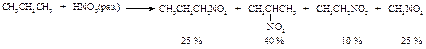
Сульфохлорирование:
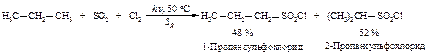
В реакциях сульфоокисления и сульфохлорирования замещению не подвергаются атомы водорода при третичном углероде из-за пространственных затруднений для подхода реагента с большим объемом к третичному атому углерода.
Методы синтеза алканов
Алканы можно получать в практически неограниченном количестве из природного газа и нефти. Однако выделение индивидуальных углеводородов с увеличением в них числа атомов углерода является трудной задачей, так как при этом резко возрастает число изомерных соединений и одновременно уменьшаются различия в их физических свойствах. Поэтому для получения индивидуальных углеводородов используются синтетические методы.
Гидрирование галогеналканов
При каталитическом гидрировании галогеналканов в присутствии палладия образуются алканы.
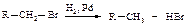
Для восстановления галогеналканов можно использовать также цинк в соляной кислоте и натрий в спирте. Иодалканы могут быть восстановлены нагреванием в запаянной ампуле с иодоводородом.
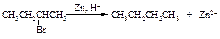
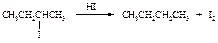
Реакция Вюрца
При взаимодействии первичных галогеналканов с металлическим натрием образуются алканы с удвоенным числом атомов углерода. Эта реакция пригодна, прежде всего, для получения высших алканов симметричного строения.
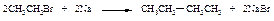
В случае использования в качестве исходных соединений различных галогеналканов в результате реакции получается смесь трех углеводородов:
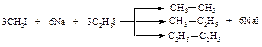
Эту смесь приходится разделять, что не всегда возможно.
Вместо натрия в этой реакции могут быть использованы и другие металлы, например магний, цинк, литий.
Реакционная способность галогеналканов уменьшается в ряду: R–I > R–Br > R–Cl.
Синтез Кольбе
При электролизе натриевых и калиевых солей карбоновых кислот образуются углеводороды симметричного строения.
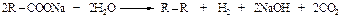
На первой стадии происходит анодное окисление анионов кислот до радикалов RСОО·, которые отщепляют СО2, а затем димеризуются. На катоде образуется водород и гидроксид щелочного металла.
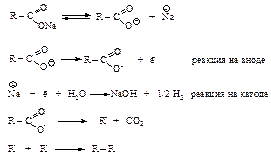
Метод Фишера-Тропша
Каталитическое гидрирование окиси углерода протекает в присутствии катализатора, содержащего кобальт или железо, с образованием смеси алканов с небольшой молекулярной массой.
Обозначение конфигураций
Для этой цели наиболее широко используют символы R и S . Эта система обозначений предложена Р. Каном (Химическое общество, Лондон), К. Ингольдом (Университетский колледж, Лондон) и В. Прелогом (Федеральная высшая техническая школа, Цюрих).
Согласно этой системе, сначала определяют старшинство, или последовательность, заместителей, т. е. четырех атомов или групп, связанных с асимметрическим атомом углерода, исходя из правила старшинства.
Правило старшинства 1. Если с асимметрическим атомом углерода связаны четыре различных атома, то старшинство зависит от атомного номера, причем более старшим будет атом с большим атомным номером. Если два атома являются изотопами одного элемента, то преимущество имеет атом с большим массовым числом. Например, в хлориодметансульфокислоте атомы, согласно их старшинству, располагаются в следующей последовательности: I > С1 > S > Н; в a-дейтероэтилбромиде – Вг > С > О > Н.
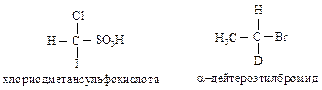
Правило старшинства 2. Если относительное старшинство групп нельзя определить с помощью правила 1, то необходимо провести аналогичное сравнение для следующих атомов в группах (и так далее, если необходимо, двигаясь дальше от асимметрического атома углерода). Иначе говоря, если асимметрический атом углерода связан с одинаковыми атомами, то следует сравнить заместители, связанные с каждым из этих первых атомов. Например, рассмотрим втор-бутилхлорид, в котором с асимметрическим атомом углерода связаны два углеродных атома. В СН3-группе следующими атомами являются Н, Н и Н; в С2Н5-группе – С, Н, Н.
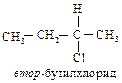
Поскольку углерод имеет больший атомный номер, чем водород, то С2Н5 старше. Таким образом, во втор-бутилхлориде заместители, согласно своему старшинству, располагаются следующим образом: С1 > С2Н5 > СН3 > Н.
В З-хлор-2-метилпентане атомы С, С и Н изопропильной группы старше С, Н и Н этильной группы и полная последовательность заместителей будет следующей: С1 > изопропил > этил > Н.
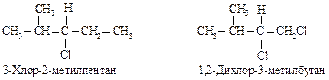
В 1,2-дихлор-З-метилбутане группа СН2Cl старше (С1, Н, Н) изопропильной (С, С, Н). Хлор имеет больший атомный номер, чем углерод, и то, что имеется два атома углерода и только один С1, не имеет значения. (Один больший номер значит больше, чем два или три меньших.)
Правило старшинства 3. Атом, связанный двойной или тройной связью, считается соответственно за два или три атома. Таким образом,
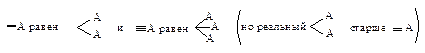
Например, в глицериновом альдегиде ОН-группа является старшей; СНО (О, О, Н) старше СН2ОН (О, Н, Н). Полная последовательность заместителей будет –ОН > –СНО > –СН2ОН > –Н.
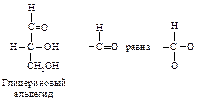
Фенильная группа С6Н5 рассматривается в виде одной из структур Кекуле:
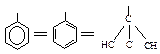
В 1-амино-2-метил-1-фенилпропане, например, фенильная группа (С, С, С) старше изопропильной (С, С, Н), но младше, чем N, который имеет больший атомный номер. Последовательность будет NН2 > С6Н5 > С3Н7 > Н.
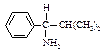
[Согласно правилу, оба атома кратной связи удваиваются (или утраиваются), так что С=О становится 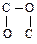 . Это более простое, но менее точное правило достаточно для рассматриваемых примеров.]
. Это более простое, но менее точное правило достаточно для рассматриваемых примеров.]
Например, в случае СНСlВгI с асимметрическим атомом углерода связаны четыре различных атома, и старшинство их зависит только от атомного номера, причем, чем больше атомный номер, тем старше заместитель. Таким образом, в порядке уменьшения их старшинства атомы располагаются в следующем порядке: I > Вг > С1 > Н.
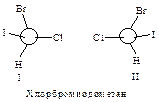
Затем молекулу располагают, так, чтобы младшая группа была направлена от наблюдателя, и рассматривают расположение оставшихся групп. Если старшинство этих групп уменьшается по часовой стрелке, то конфигурацию обозначают символом R (от латинского rectus – правый); если же старшинство этих групп уменьшается против часовой стрелки, то конфигурацию обозначают символом S (от латинского sinister – левый).
Таким образом, конфигурации I и II выглядят следующим образом:
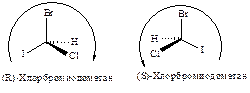
и обозначаются соответственно символами R и S.
Полное название оптически активного соединения отражает и конфигурацию и направление вращения, как, например, (S)-(+)-втор-бутилхлорид. Рацемическую модификацию можно обозначить символом R , S, например (R , S)-втор-бутилхлорид.
Конечно, нельзя путать направление оптического вращения соединения (такого же физического свойства реального вещества, как температура кипения или плавления) с направлением нашего взгляда, когда мы мысленно располагаем молекулу каким-то определенным условным образом. Пока для определенного соединения экспериментально не установлена связь между конфигурацией и знаком вращения, нельзя сказать, знак (+) или (–) соответствует (R)- или (S)-конфигурации.
ЦИКЛОАЛКАНЫ
Углеводороды, которые содержат кольца, состоящие из углеродных атомов, связанных между собой простыми связями, называются алициклическими или циклоалканами (циклопарафинами). Ниже приведена полная и сокращенная запись структурных формул первых четырех представителей ряда циклоалканов.
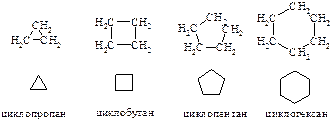
Номенклатура. Изомерия
Названия циклоалканов образуются добавлением приставки цикло- к названию линейного алкана с тем же числом атомов углерода. В алициклических соединениях известны следующие виды изомерии: структурная (изомерия, связанная с различной величиной цикла, различным строением и положением в цикле боковых цепей), пространственная (геометрическая или цис-, транс-изомерия, обусловленная различным расположением групп относительно плоскости кольца) и оптическая (энантиомерия).
Примеры структурных изомеров C6H12:
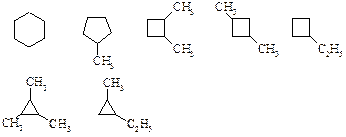
Примеры геометрических изомеров:
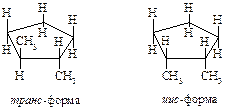
Примеры энантиомеров:
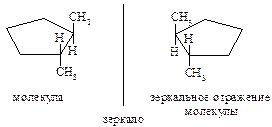
Физические свойства
Физические свойства циклоалканов сходны со свойствами соответствующих ациклических углеводородов, хотя температуры кипения и плавления циклических соединений немного выше. В частности, малые циклоалканы – циклопропан и циклобутан – бесцветные газы, не имеющие запаха, а циклопентан и циклогексан – бесцветные жидкости с Ткип. 50,5 и 80 оС соответственно. Циклоалканы неполярные или малополярные соединения, поэтому они хорошо растворимы в неполярных растворителях, таких как четыреххлористый углерод, эфир, и нерастворимы в сильно полярном растворителе - воде.
Типы напряжения
Несмотря на то, что и алканы и циклоалканы образованы атомами углерода, находящимися в одном и том же состоянии гибридизации (sp 3 ), циклоалканы имеют ряд структурных особенностей. Указанные особенности связаны прежде всего с напряжением молекулы при циклообразовании.
Угловое напряжение (напряжение Байера) – увеличение энергии молекулы, вызванное отклонением угла между связями от величины нормального тетраэдрического угла (109о28′).
Торсионное напряжение (напряжение Питцера, напряжение заслоненных связей) – увеличение энергии, вызванное отклонением конформации любого этанподобного звена в молекуле циклоалкана от заторможенной.
Трансаннулярное напряжение (напряжение Прелога) - увеличение энергии молекулы вследствие взаимодействия несвязанных атомов и фрагментов (двойных связей, функциональных групп и т.д.); такой тип взаимодействия носит также название «взаимодействие через пространство, цикл»
Строение
Теория напряжения А. Байера. В 1885 г. профессор Мюнхенского университета А.Байер предложил теорию, объясняющую некоторые аспекты химии циклических соединений. Часть его теории, рассматривающая способность к раскрытию малых циклов, общепринята и сегодня, хотя сейчас она излагается с других, современных позиций.
Байер рассуждал следующим образом. Когда атом углерода связан с четырьмя другими атомами (sp3-гибридизация), между каждыми двумя связями образуется угол 109о28¢.
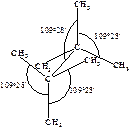
Предполагалось, что в молекулах циклоалканов атомы углерода являются вершинами правильных плоских многоугольников. Циклопропан представляет собой плоский правильный треугольник с углом между связями С–С, равным 60о.
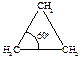
В циклопропане две связи у каждого из атомов углерода не могут образовать нормальный тетраэдрический угол 109о28¢, угол между ними сжат до 60о. Такое отклонение от нормального тетраэдрического угла делает эту молекулу “напряженной” и, следовательно, неустойчивой по сравнению с молекулами с тетраэдрическими углами.
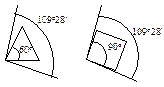
Циклоропан вступает в реакции с раскрытием кольца, поскольку при этом снимается угловое напряжение и образуются более устойчивые ациклические соединения. Чем больше отклонение от нормального угла 109о28¢, тем более “напряженной” является молекула: для циклопропана отклонение составляет 1/2(109о28¢ - 60о)= 24о44¢, а для плоского циклобутана -1/2(109о28¢ - 90о)= 9о44¢.
Поскольку искажение углов наиболее значительно в циклопропане, то он является более “напряженным”, более неустойчивым, более склонным к реакциям, протекающим с раскрытием кольца. Углы в правильном плоском пятиугольнике весьма близки к тетраэдрическим (108о), и поэтому циклопентан практически свободен от углового напряжения.
Углы в правильном плоском шестиугольнике (120о) несколько превышают тетраэдрические, на основании чего Байер предположил (ошибочно), что в циклогексане должно быть некоторое напряжение, а при переходе к циклогептану, циклооктану и т. д. отклонения от угла 109о28¢ будут увеличиваться, вследствие этого молекулы будут становиться все более напряженными.
Как согласуется теория Байера с фактами?
Полезную информацию об относительной устойчивости органических соединений дают теплоты сгорания веществ. Теплота сгорания - это количество теплоты, которое выделяется при сгорании одного моля вещества до улекислого газа и воды. Рассчитано, что для алифатических соединений сгорание метиленового звена -СН2- дает 659,0 кДж×моль -1 .
-СН2- + О2 ® СО2 + Н2О + тепло
В основе классификации циклов, первоначально кажущейся произвольной, лежит зависимость между размером кольца и его устойчивостью.
Если циклопропан и циклобутан выделяют при сгорании больше энергии в расчете на СН2-группу, чем ациклические соединения (соответственно на 38,5 и 27,7 кДж/моль), то это означает, что они содержат больше энергии (см. табл. 7.1). Тогда в соответствии с теорией напряжения Байера циклопропан и циклобутан менее устойчивы по сравнению с ациклическими соединениями, и это обусловливает их склонность к реакциям с раскрытием кольца. В соответствии с теорией Байера циклы большие, чем циклогексан и циклобутан, должны быть неустойчивыми и иметь высокие теплоты сгорания. Но из таблицы 7.1 видно, что теплоты сгорания нормальных и средних циклов в расчете на одну СН2-группу мало отличаются от теплоты сгорания СН2-группы ациклических углеводородов, а для больших циклов - теплота сгорания СН2-группы практически равна этой величине. В противоположность теории Байера ни одна из этих систем не обладает меньшей устойчивостью по сравнению с ациклическими соединениями и не обнаруживает тенденции вступать в реакции раскрытия цикла подобно циклопропану.
Что же неверно в теории Байера? Только одно: Байер считал, что кольцо является плоским, но только трехчленный цикл должен быть плоским. Циклы, содержащие большее число атомов углерода, не обладают плоской конфигурацией.
Что же означает угловое напряжение с позиций современной теории? Для образования связей необходимо такое расположение атомов, при котором орбитали одного атома перекрывались бы с орбиталями другого атома. Чем больше такое перекрывание, тем прочнее связь. Когда углерод связан с четырьмя другими одинаковыми атомами, его sp3-гибридные орбитали направлены к углам тетраэдра. Образование связи происходит в результате перекрывания его sp3-орбитали с аналогичной sp3-орбиталью другого атома. Такое перекрывание наиболее эффективно и связь наиболее прочна, если два атома расположены так, что оси этих sp3-гибридных орбиталей лежат на прямой, соединяющей ядра атомов. В этом случае угол между углерод-углеродными связями С-С-С должен составлять 109о28¢ (рис. 7.1).
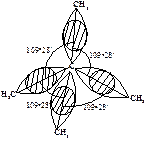
Рис. 7.1. Молекула алкана. Оси sp 3-гибридных облаков лежат на линии, соединяющей ядра атомов
Таблица 7.1
Химические свойства
Химические свойства циклоалканов во многом совпадают со свойствами алканов. Для них характерны прежде всего реакции радикального замещения.
Помимо реакций свободнорадикального замещения, характерных для циклоалканов, циклопропан и циклобутан вступают в некоторые реакции присоединения с раскрытием кольца.
Гидрирование. Циклопропан реагирует с водородом в присутствии катализатора (Ni, T = 80 оС) с разрывом кольца.
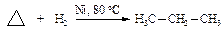
Циклобутан также взаимодействует с разрывом цикла, но при более высокой температуре 200 оС.
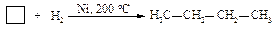
Пятичленный цикл разрывается только при значительно более высокой температуре 300 оС.
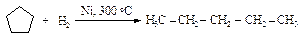
Циклогексан в этих условиях дегидрируется, кольцо сохраняется.
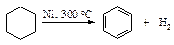
Галогенирование. Реакция с бромом также идет по-разному в зависимости от размера цикла.
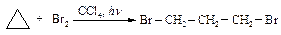
Циклобутан не взаимодействует с бромом подобным образом.
Циклопентан и циклогексан реагируют с галогенами (Cl2, Br2) по механизму радикального замещения.
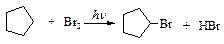
Гидрогалогенирование. Циклопропан взаимодействует с иодоводородом, как ненасыщенное соединение - присоединяет галогеноводород, при этом происходит раскрытие цикла.
Остальные циклопарафины с галогеноводородами не реагируют.
В реакциях циклоалканов проявляется различие в свойствах малых циклов и пяти-, шестичленных циклов. Циклопропан, а также циклобутан (в меньшей степени) вступают в реакции присоединения, проявляя свойства ненасыщенных соединений.
Способы получения
Диеновый синтез
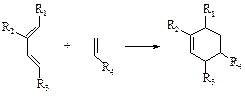
Алкены
Углеводороды состава СnН2n с открытой цепью, содержащие одну двойную связь, называются алкенами. Простейшим углеводородом этого ряда является этилен СН2=СН2. Атом углерода в этилене находится в sp2-гибридном состоянии (тригональный углерод). За счет трех гибридизованных орбиталей каждый атом углерода образует три s-связи: одну - с соседним атомом углерода, две - с двумя атомами водорода. Боковое перекрывания двух 2р-орбиталей атомов углерода дает p-связь и делает невозможным вращение вокруг s-связи углерод-углерод. Этим обусловлено явление геометрической изомерии.
Геометрические изомеры (состав и способ связывания атомов одинаков, расположение групп и атомов в пространстве различно). Для названия этих изомеров используется Е,Z-номенклатура. При этом возможно использование классических цис- и транс-обозначений для определения пространственного расположения одинаковых или сходных групп относительно плоскости сравнения.
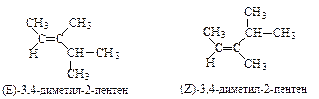
| Одинаковые по старшинству группы расположены по разные стороны от плоскости двойной связи. В этом соединении два метильных радикала находятся в цис-положении. | Одинаковые по старшинству группы расположены по одну сторону от плоскости двойной связи. В этом соединении два метильных радикала находятся в транс-положении. |
Относительное старшинство заместителей при каждом атоме углерода с двойной связью определяется по атомному номеру: Н (атомный номер - 1) - младший, С (атомный номер - 6) - старший заместитель; если атомы при углероде с двойной связью одинаковы, то рассматривается старшинство последующих атомов: - СН3 (последующие атомы - Н, Н, Н) - младший заместитель; -СН(СН3)2 (последующие атомы - Н, С, С) - старший заместитель.
Физические свойства
Физические свойства алкенов в основном сходны с соответствующими свойствами алканов. Алкены нерастворимы в воде, но хорошо растворимы в неполярных растворителях, таких как бензол, тетрахлорметан. Их плотность меньше плотности воды. Так же как и в случае алканов, температуры кипения повышаются на 20 - 30 оС при увеличении длины цепи на один атом углерода (за исключением низших алкенов). Разветвление углеродной цепи в молекулах изомеров понижает температуру кипения (табл. 8.1).
цис-Изомер менее симметричен, чем транс-изомер, поэтому его упаковка в кристаллической решетке менее плотная, что обусловливает, как правило, более низкую температуру плавления цис-изомера.
Алкильная группа подает электроны к углероду с двойной связью. Это объясняется разной гибридизацией атомов углерода. Углерод в состоянии sp 2-гибридизации сильнее притягивает электроны, чем углерод в состоянии sp 3-гибридизации. Поэтому связь поляризована.
Таблица 8.1
Физические свойства алкенов
| Название | Формула | Тпл, ºС | Ткип, ºС | Плотность, г/см3 |
| Этен | СН2=СН2 | -169 | -102,0 | – |
| Пропен | CH2=CH–CH3 | -188 | -48,0 | – |
| 1-Бутен | СН2=СН–СH2–СН3 | -185 | -6,5 | – |
| 1-Пентен | СН2=СН–(СН2)2–СН3 | -185 | 30,0 | 0,643 |
| 1-Гексен | СН2=СН–(СН2)3–СН3 | -138 | 63,5 | 0,675 |
| 1-Гептен | СН2=СH–(СН2)4–СН3 | -119 | 93,0 | 0,698 |
| цис-2-Бутен | 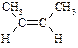
| -139 | 4,0 | – |
| транс-2-Бутен | 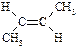
| -106 | 1,0 | – |
| Метилпропен | CH2=C(CH3)2 | -141 | -7,0 | – |
Химические свойства
Атом или группа атомов, которая определяет свойства какого-либо класса органических соединений, называется функциональной группой. В алкенах функциональной группой является двойная углерод-углеродная связь. В алкене более сложном, чем этилен, присутствуют алкильные группы. В определенных условиях алкильные группы в этих молекулах могут вступать в реакции, типичные для алканов. Однако характерными реакциями алкенов являются реакции по двойной углерод-углеродной связи.
Когда имеется сложная молекула с несколькими функциональными группами, то можно ожидать, что свойства этой молекулы будут сочетать свойства различных функциональных групп. Однако свойства отдельной группы будут несколько изменяться под влиянием других групп, и важно понимать эти изменения.
Двойная связь состоит из прочной s-связи и менее прочной p-связи. Типичными реакциями двойной связи являются реакции, в которых происходит разрыв менее прочной p-связи и образование вместо нее двух более прочных s-связей. Такие реакции называются реакциями присоединения. Они обозначаются символом Аd ( Addition - присоединение).
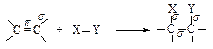
Какие реагенты могут присоединяться к двойной углерод-углеродной связи? Для ответа рассмотрим электронное строение алкенов. Облака p-электронов находятся над и под плоскостью, в которой лежат атомы углерода и водорода. Эти электроны наиболее доступны для реагентов с недостатком электронов. Двойная углерод-углеродная связь служит донором электронов, т.е. ведет себя как основание*. Она реагирует с соединениями, которые обеднены электронами, т.е. с кислотами. Эти реагенты, не имеющие пары электронов, называются электрофильными реагентами (электрофил - любящий электроны). Следовательно, типичными реакциями алкенов являются реакции электрофильного присоединения (AdE).
Существуют реагенты другого типа, также обедненные электронами - свободные радикалы. С ними алкены вступают в реакции радикального присоединения (AdR) .
Большинство алкенов содержит алкильные группы, которые являются остатками алканов, следовательно, такие алкены могут подобно алканам участвовать в реакциях свободнорадикального замещения атома водорода в алкильном остатке (SR).
Реакция электрофильного присоединения протекает в две стадии:
I стадия - медленная, присоединение электрофила ЕÅ с образованием карбокатиона.
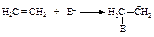
II стадия - быстрая, присоединение нуклеофила Nu (нуклеофил - любящий ядро).
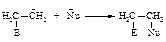
По такому механизму протекает реакция присоединения галогеноводородов НСl, НВr, НI.
Для присоединения воды к алкенам необходим катализатор - сильная минеральная кислота, которая дает протон НÅ - электрофил, а нуклеофилом является вода (Н2О) за счет неподеленной пары электронов на атоме кислорода. Подобным образом с алкенами реагирует спирт (RОH), который также имеет неподеленную электронную пару на атоме кислорода. В реакции с серной кислотой быстрая стадия - взаимодействие с нуклеофилом —ОSO2OH.
Присоединению хлора и брома, молекулы которых неполярны, предшествует поляризация
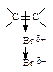
Под влиянием электронного облака двойной углерод-углеродной связи изменяется распределение электронной плотности в молекуле галогена. В первой медленной стадии присоединяется электрофил Br+, во второй - нуклеофил Br—.
Частицы, присоединяющиеся в медленной и быстрой стадиях электрофильного присоединения, приведены в таблице 4.2.
Таблица 4.2.
Реакции электрофильного присоединения
| Реакции присоединения | |||||
| Реагент | галогенов | галоген- водородов | воды | спиртов | серной кислоты |
| Электрофил | Cl+ Br+ | H+ | H+ | H+ | H+ |
| Нуклеофил | Cl— Br— | Cl— , Br—, I— | Н2O | ROH | — ОSO2OH |
Присоединение галогенов
Алкены реагируют с бромом и хлором в инертном растворителе (например, в четыреххлористом углероде), образуя вицинальные дигалогениды (vicinalis - соседний).
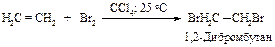
Этот процесс протекает как электрофильное присоединение.
Механизм реакции
Первая стадия. Неполярная молекула галогена поляризуется под действием богатой электронами двойной углерод-углеродной связи. Изменение распределения электронной плотности в одной молекуле под влиянием внешних условий (растворитель, другие молекулы, ионы) называется поляризацией. Поляризованная таким образом молекула галогена взаимодействует с p-cистемой, образуя p-комплекс:
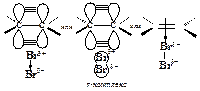
Вторая стадия. p-Комплекс разрушается, ион брома ВrÅ- электрофил - присоединяется к атому углерода и образуется s-комплекс - карбокатион. Вторая стадия - медленная, определяет скорость всего процесса.
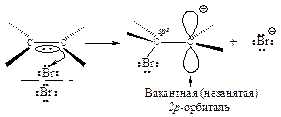
Положительно заряженный атом углерода находится в sр2-гибридизованном состоянии (его 2р-орбиталь не занята). Свободная 2р-орбиталь перекрывается с неподеленной парой электронов атома брома, образуя циклический ион бромония.
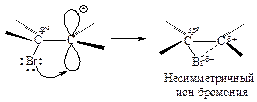
Третья стадия. Оставшийся анион брома (нуклеофил - ядро любящий) атакует образовавшийся циклический ион бромония по второму атому углерода.
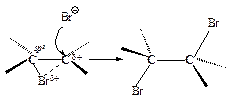
Стадия 2 – медленная, с высокой энергией активации
Доказательствами справедливости этого механизма служат следующие факты:
1). При проведении бромирования в растворе, содержащем помимо аниона брома другой нуклеофил (например, анион хлора), в продуктах реакции появляется наряду с дибромидом соединение, содержащее хлор.
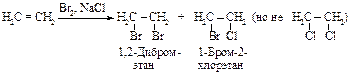
8 .2.1.2. Реакционная способность галогенов в реакции А d Е
Присоединение хлора и брома к алкенам происходит легко и во многих случаях с количественным выходом. Присоединение йода к алкенам осуществить не удается. Взаимодействие фтора с алкенами происходит так энергично, что углеводород распадается на осколки с меньшим числом атомов углерода.
Механизм реакции
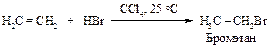
Алкены реагируют с хлористым, бромистым (в отсутствие пероксидов) или йодистым водородом по механизму электрофильного присоединения. Реакцию проводят, пропуская газообразный галогеноводород непосредственно в алкен, используя растворитель, который растворяет и неполярный алкен и полярный галогеноводород, например, ССl4.
Присоединение галогеноводорода протекает в две стадии.
Первая стадия. Присоединение электрофила - протона НÅ к алкену с образованием карбокатиона.
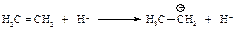
Вторая стадия. Взаимодействие нуклеофила Br- с карбокатионом.

Медленной стадией является первая - присоединение электрофила с образованием карбокатиона. Она определяет общую скорость присоединения и тип реакции - электрофильное присоединение.
Порядок увеличения реакционной способности галогеноводородов по отношению к алкенам совпадает с порядком увеличения их кислотности: HF << HCI < HBr < HI. Это еще раз подтверждает положение о том, что лимитирующей стадией является атака двойной связи протоном.
Перегруппировка
Реакции электрофильного присоединения протекают через стадию образования карбокатионов и могут сопровождаться перегруппировками.
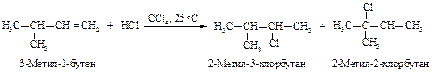
Механизм реакции
Реакция начинается с присоединения электрофила НÅ с образованием карбокатиона.
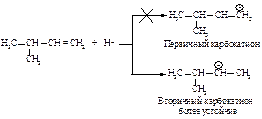
Образовавшийся устойчивый вторичный карбокатион может перегруппировываться в более устойчивый третичный карбокатион. Перегруппировка состоит в переносе гидрид-иона НΘ (протон с двумя электронами) к соседнему атому углерода (1,2-сдвиг).
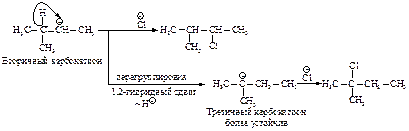
Аналогичным образом может происходить и миграция карбаниона :CH3Ө от соседнего четвертичного атома углерода.
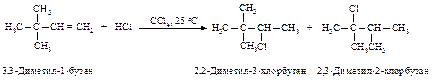
Механизм реакции
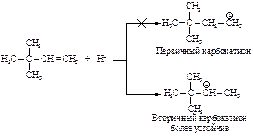
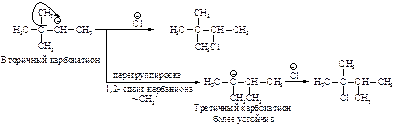
Перегруппировка происходит в том случае, если при 1,2-сдвиге гидрид-иона или отрицательно заряженной CH 3 -группы может образоваться более устойчивый карбокатион.
Поскольку перегруппировки происходят только в карбокатионах, а карбанионы и радикалы не подвергаются перегруппировкам, то можно утверждать: если в результате реакции образуется продукт с иным углеродным скелетом, чем в исходной молекуле, или присоединение происходит не у того атома углерода, у которого была двойная связь, то можно предположить, что реакция протекает с участием карбокатиона в качестве промежуточной частицы.
Пероксидов
В отсутствие пероксидов взаимодействие бромоводорода с алкенами протекает как электрофильное присоединение в соответствии с правилом Марковникова. В присутствии пероксидов алкены реагируют с бромоводородом по другому механизму, и поэтому направление присоединения не соответствует правилу Марковникова.
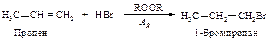
Для того чтобы объяснить перекисный эффект, М. Хараш и Ф. Майо предположили, что присоединение бромоводорода к алкенам может происходить по двум совершенно различньм механизмам: присоединение в отсутствие пероксида - по ионному механизму, а присоединение в присутствии пероксида - по цепному свободнорадикальному.
Механизм реакции
1. Инициирование:
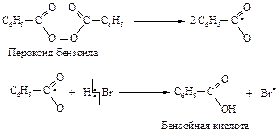
2. Рост цепи:
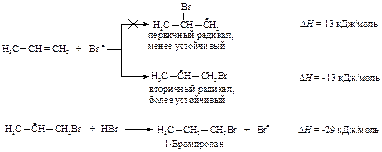
Свободные радикалы, как и катионы, являются электронодефицитными частицами. Алкен предоставляет электроны p-системы для завершения электронной оболочки атома брома. Атом брома присоединяется к алкену и превращает его в свободный радикал. Образовавшийся при этом более устойчивый вторичный углеводородный радикал отщепляет атом водорода от бромоводорода. Теперь присоединение завершено, образовался новый атом брома, чтобы продолжить цепь.
3. Обрыв цепи:
Присоединение бромводорода в присутствии пероксида происходит формально против правила Марковникова потому, что в медленной стадии с алкеном реагирует атом брома.
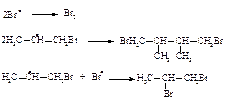
Медленная стадия радикального присоединения бромоводорода в присутствии пероксида:
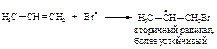
Медленная стадия электрофильного присоединения бромоводорода в отсутствие пероксида:
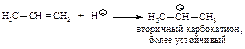
Таким образом, реакции свободнорадикального и электрофильного присоединения подчиняются одному и тому же правилу.
Гидроборирование алкенов
Боран BH3 легко присоединяется к алкенам, образуя триалкилборан. Использование формулы борана BH3 вместо формулы диборана B2H6 (именно в виде такого соединения существует боран) вполне корректно, т.к. в растворителях, представляющих собой основание Льюиса, диборан существует в виде донорно-акцепторного комплекса (ДАК):
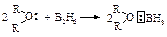
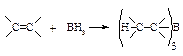
В случае несимметричного алкена бор присоединяется к наиболее гидрогенизированному атому углерода при двойной связи.
Атом бора, имеющий только шесть электронов, атакует углерод, при этом p-электроны двойной связи смещаются к атому бора.
Направление присоединения объясняется большей устойчивостью переходного состояния (ПС1) по сравнению с переходным состоянием (ПС2):
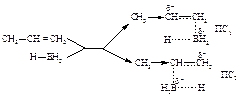
В отличие от электрофильного присоединения реакция не протекает через стадию образования карбониевого иона. Основной движущей силой реакции является присоединение бора к углероду: в переходном состоянии связь C–B образуется легко и раньше, чем связь C–H, поэтому на атоме углерода должен возникнуть некоторый положительный заряд, который в ПС1 распределен в большей степени, чем в ПС2. По мере того, как атом углерода теряет p-электроны и становится электронодефицитным, он начинает присоединять ион водорода с парой электронов, связанный с бором. Бор отдает этот водород с парой электронов, поскольку сам приобретает p-электроны алкена.
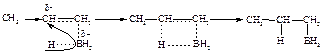
Алкилбораны при окислении гидропероксидом в присутствии щелочи превращаются в спирты, которые нельзя получить реакцией гидратации алкенов.
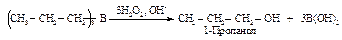
Алкилирование алкенов
Присоединение насыщенного углеводорода к алкену называется алкилированием и используется в нефтехимической промышленности. Наибольшее значение имеет присоединение изобутана к изобутену в присутствии минеральных кислот, приводящее к 2,2,4-триметилпентану – изооктану.
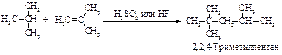
Первая стадия - такая же, как и во всех реакциях электрофильного присоединения. Электрофил (протон) присоединяется в соответствии с правилом Марковникова с образованием трет-бутил-катиона (I).
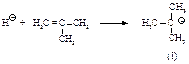
Образующийся карбокатион (I) аналогичным образом атакует вторую молекулу алкена с образованием нового карбокатиона (II).
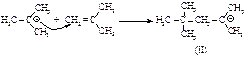
Карбокатион (II) может взаимодействовать с изобутаном, имеющим третичный атом водорода: карбокатион (II) вырывает третичный водород с его парой электронов - гидрид-ион, при этом образуется карбокатион (I) и изооктан.
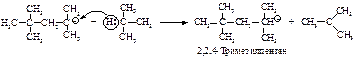
Следовательно, механизм алкилирования изобутена изобутаном состоит в следующем: сначала происходит димеризаця изобутена под действием серной кислоты как катализатора, после чего карбокатион (II) вступает в реакцию переноса гидрид-иона с изобутаном.
Окисление
Окисление без разрыва углерод-углеродной связи.
Характер окисления алкенов зависит от используемого окислителя и условий реакции. Действие окислителя, который является электрофилом, всегда направлено в место повышенной электронной плотности - на атомы углерода, связанные двойной связью. При действии холодного раствора перманганата калия (слабый окислитель) разрывается только p-связь и образуется две s-связи с кислородом (реакция Вагнера).
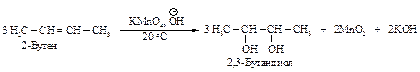
Каталитическое окисление алкенов над серебром приводит к алкоксидам.
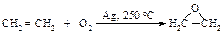
Действие другого мягкого окислителя - надкислоты - также приводит к образованию алкоксидов (реакция Н. А. Прилежаева).
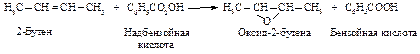
Окисление с разрывом углерод-углеродной связи.
Под влиянием сильного окислителя ( КМnO4, НÅ ) разрывается не только p-, но и s-связь, разрушается углеродный скелет. В продуктах окисления все связи атомов углерода фрагмента >С=С<, кроме связей с соседними алкильными группами, насыщены кислородом:
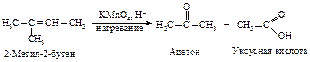
Концевая группа =СН2 окисляется до оксида углерода и воды.
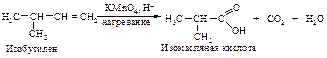
Озонолиз (озонирование и последующий гидролиз озонида) проводится в две стадии: присоединение озона по двойной связи с образованием озонида и гидролиз полученного озонида в присутствии восстанавливающего агента, например, цинковой пыли. Восстановитель необходим для связывания пероксида водорода, который может реагировать с альдегидами.
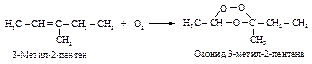
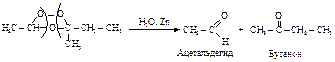
Озонолиз является методом определения структуры неизвестного алкена путём разрушения его на несколько меньших фрагментов, которые легко идентифицируются. Зная строение полученных альдегидов и кетонов, можно установить структуру исходного алкена.
Окисление алкенов в присутствии солей палладия (Вакер-процесс). Алкены окисляются кислородом в присутствии солей палладия и меди.
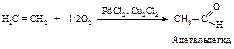
Этот способ является промышленным способом получения ацетальдегида. Окисление других алкенов идет по наименее гидрогенизированному атому углерода.
Полимеризация алкенов
Полимер - это макромолекула, состоящая из очень большого числа повторяющихся звеньев. Он образуется путем последовательного присоединения малых молекул, называемых мономерами.
Полимер, получаемый из одинаковых мономеров, называется гомополимером, а полученный из двух различных мономеров - сополимером или гетерополимером.
| nA ® –A–A–A–A– или An |
| гомополимер |
| mB + nA ® –A–B–B–A–B–A–A–B–B–A– |
| сополимер (случайное расположение) |
Полимер представляет собой смесь молекул с разной молекулярной массой, которая колеблется в определенных пределах. Полимеры содержат "концевые группы", которые отличаются от повторяющихся звеньев.
|
| |||
| Инициатор полимеризации |
концевые группы | ||

 X––Y + nA ® X––(An)––Y
X––Y + nA ® X––(An)––YКонцевые группы представляют собой незначительную часть полимера, поэтому их характер не учитывается при рассмотрении свойств полимера.
Катионнаяполимеризация
Катионная полимеризация протекает под действием кислот Льюиса (Н+, А1С13, BF3) по механизму, сходному с механизмом электрофильного присоединения по двойной связи. На первой стадии протон (или другая кислота) присоединяется к алкену, образуя карбокатион.
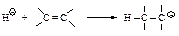
Затем другая молекула алкена вступает в реакцию за счет электронной пары двойной связи, и образуется карбокатион с более длинной цепью.
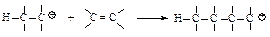
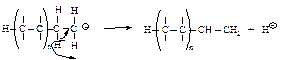
Далее процесс многократно повторяется и образуется карбокатион с высокой молекулярной массой. Эта цепь, в конце концов, может оборваться из-за разрушения катионного центра по какой-либо причине, например, из-за отрыва протона.
Изобутилен легко полимеризуется по катионному механизму, поскольку содержит электронодонорные алкильные группы.
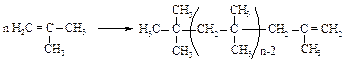
Цепь полиизобутилена формируется таким образом, что на ее конце образуется наиболее стабильный карбокатион. Поэтому присоединение происходит упорядоченно - "голова к хвосту".
В условиях, близких к безводным, образуется полимер с очень длинной цепью. В присутствии серной кислоты с массовой долей 60 % изобутилен дает смесь димеров. Обрыв цепи происходит после соединения двух молекул изобутилена, промежуточный карбокатион теряет протон, отдавая его воде прежде, чем успеет прореагировать со следующей молекулой алкена. Вода выступает в роли основания, забирая протон у карбокатиона димера.
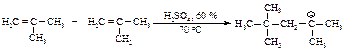
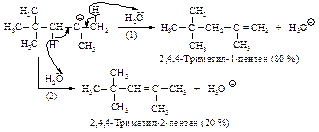
Можно ожидать, что 2,4,4-триметил-2-пентен благодаря центральному расположению двойной связи более устойчив, чем 2,4,4-триметил-1-пентен, имеющий концевую двойную связь. Однако в данном конкретном случае наличие объёмистой трет-бутильной и метильной групп в цис-положении у соседних атомов углерода создаёт пространственные затруднения образованию алкена:
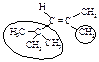
Гидрирование 2,4,4-триметил-1-пентена и 2,4,4-триметил-2-пентена приводит к 2,2,4-триметилпентану (изооктану), используемому в качестве топлива для бензиновых двигателей внутреннего сгорания. Для изооктана принята стандартная антидетонационная характеристика (октановое число), равная 100 (октановое число н-гептана - 0).
Анионная полимеризация.
При анионном механизме полимеризация алкена инициируется атакой нуклеофильного реагента (NaNH2, NaOC4H9-н) по одному из концов двойной связи.
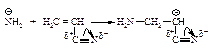
В этом примере амид-анион NH2– - нуклеофил атакует двойную связь акрилонитрила, возникающий карбанион стабилизирован делокализацией отрицательного заряда между атомом углерода и соседней электроноакцепторной группой –Сº N.
Если промежуточный анион окажется достаточно устойчивым, происходит его присоединение к другой молекуле алкена.
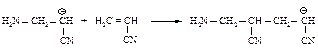
Растущая цепь может оборваться при любой реакции, которая приведёт к исчезновению отрицательного заряда на конце цепи.
Простые алкены, не имеющие электроноакцепторных заместителей, не присоединяют анионы и не образуют устойчивых карбанионов.
Свободнорадикальная полимеризация.
Этилен может полимеризоваться в присутствии пероксидов при высоком давлении и повышенной температуре.
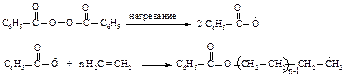
Обрыв цепи происходит вследствие рекомбинации
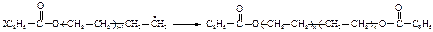
или диспропорционирования радикалов.
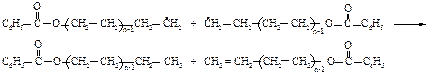
Полиэтилен содержит в углеводородной цепи 100-1000 звеньев этилена. Он обладает рядом ценных свойств и широко используется в производстве упаковочной плёнки, для изготовления большого числа изделий методом литья и формования.
При свободнорадикальной полимеризации несимметричного алкена растущий конец полимера представляет собой наиболее стабильный радикал, полимеризация идёт по типу “голова к хвосту”.
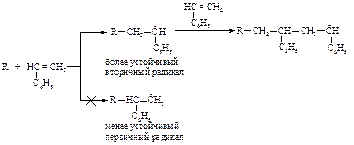
Пропилен и изобутилен не полимеризуются по свободнорадикальному механизму.
Координационнаяполимеризация осуществляется с помощью катализаторов Циглера-Натта. Наиболее распространённый из них - комплекс триэтилалюминия с хлоридом титана Al(C2H5)·TiCl4. С помощью таких катализаторов мономер внедряется в связь между металлом М и растущей полимерной цепью.
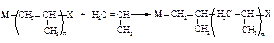
В полипропилене, который получают без этого катализатора, метильные группы расположены беспорядочно относительно основной полимерной цепи. Такой полимер называется атактическим.
Катализатор Циглера-Натта позволяет получать полипропилен, в котором метильные группы расположены упорядоченно по обеим сторонам полимерной цепи (синдиотактический).
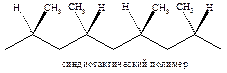
Кроме того, можно получать полипропилен, в котором все метильные группы расположены по одну сторону от основной полимерной цепи (изотактический).
Способы получения алкенов
Низшие алкены (С2 - С5), в промышленных масштабах получают из газов, образующихся при термической переработке нефти и нефтепродуктов. Алкены можно также получить, используя лабораторные методы синтеза.
Дегидрогалогенирование
При обработке галогеналканов основаниями в безводных растворителях, например, спиртовым раствором едкого кали, происходит отщепление галогеноводорода.
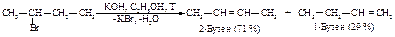
Преобладающим продуктом реакции является наиболее устойчивый, т.е. наиболее алкилированный алкен (правило Зайцева)
Дегидратация
При нагревании спиртов с серной или фосфорной кислотами происходит внутримолекулярная дегидратация (b -элиминирование).
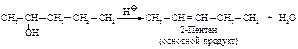
Преобладающее направление реакции, как и в случае дегидрогалогенирования, - образование наиболее устойчивого алкена (правило Зайцева).
Дегидратацию спиртов можно провести, пропуская пары спирта над катализатором (оксиды алюминия или тория) при 300 - 350 оС.
Гидрирование алкинов
При гидрировании алкинов в присутствии платинового или никелевого катализаторов, активность которых уменьшена добавлением небольшого количества соединений свинца (каталитический яд), образуется алкен, который не подвергается дальнейшему восстановлению.
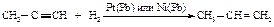
ДИЕНЫ
Диены содержат две двойные углерод-углеродные связи. Они делятся на три класса. В сопряженных диенах двойные связи разделены простой cвязью.
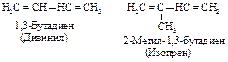
Если двойные связи разделены более чем одной простой связью, они называются изолированными.

В диенах с кумулированными двойными связями обе двойные связи находятся при одном атоме углерода.
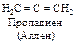
Наибольшее практическое и теоретическое значение имеют диеновые углеводороды с сопряженными двойными связями. Особенностью сопряженных диенов является высокая подвижность их p-электронной системы. Она проявляется в ряде важнейших свойств - физических и химических. Сопряженная система обнаруживается по характерным полосам поглощения в близкой к видимой ультрафиолетовой области.
Химические свойства
Способы получения
Наиболее важные сопряженные диены 1,3-бутадиен (дивинил), 2-метил-1,3-бутадиен (изопрен) получают из соответствующих алканов дегидрированием.
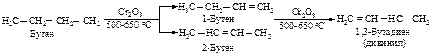
Аналогично алкенам диены могут быть получены реакцией дегидрогалогенирования дигалогенпроизводных алканов и дегидратации диолов.
1,3-Бутадиен также получают высокотемпературной каталитической реакцией дегидрирования и дегидратации этанола (процесс С.В. Лебедева).
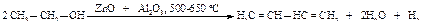
Дивинил может быть получен из ацетилена и формальдегида по методу В. Реппе (см. п. 5.3.3).
Изопрен получают по методу Г. Принса.
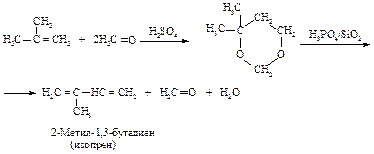
АЛКИНЫ
Алкинами называются углеводороды, содержащие тройную углерод-углеродную связь –СºС–.
Общая формула простых алкинов СnH2n-2. Простейшим представителем класса алкинов является ацетилен H–СºС–H, поэтому алкины называют также ацетиленовыми углеводородами.
Атомы углерода ацетилена находятся в sp-гибридном состоянии. При гибридизации 2s-орбитали и 2р-орбитали образуются две равноценные sp-гибридные орбитали, расположенные на одной прямой, и остаются две негибридизованные р-орбитали.
В молекуле ацетилена простая связь (s -связь) между атомами углерода образована перекрыванием двух sp-гибридизованных орбиталей. Две взаимно перпендикулярные p -связи возникают при боковом перекрывании двух пар негибридизованных 2р-орбиталей, p -электронные облака охватывают скелет так, что электронное облако имеет симметрию, близкую к цилиндрической. Связи с атомами водорода образуются за счёт sp-гибридных орбиталей атома углерода и 1s-орбитали атома водорода, молекула ацетилена линейна.
В пропине простая связь (s -связь) Сsp-Сsp3 короче аналогичной связи Сsp-Сsp2 в алкенах, это объясняется тем, что sp-орбиталь ближе к ядру, чем sp2- орбиталь .
Тройная углерод-углеродная связь С º С короче двойной связи, а общая энергия тройной связи приблизительно равна сумме энергий одной простой связи С–С (347 кДж/моль) и двух p-связей (259·2 кДж/моль) (табл. 10.1).
Таблица 10.1
Соединение
Связи между атомами С
Связи С–Н
СºС
ºС–С
Н–Сº
Сsp3–Н
Особый интерес представляет связь ºС-Н. Эта связь короче и прочнее аналогичных связей углерод-водород в алкенах и алканах (табл. 10.2).
Таблица 10.2
Характеристики связей С-Н в алканах, алкенах и алкинах
| Углеводород | Длина, l×102, нм | Энергия Е, кДж/моль | Доля s-орбитали атома углерода, % | Дипольный момент m, Д |
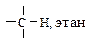
| 11,0 | 405 | 25,0 | 0,3 |
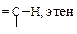
| 10,9 | 435 | 33,3 | 0,6 |
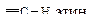
| 10,6 | 464 | 50,0 | 1,08 |
Связь ºС–Н имеет заметную полярность, связанную с большей долей s-состояния гибридной орбитали и, вследствие этого, с большей близостью электронной пары связи ºС–Н к углероду.
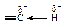
В результате смещения электронного облака s-связи от атома водорода к атому углерода атом водорода становится отчасти положительно заряженным - кислотным.
Энергия диссоциации связи углерод-водород в ацетилене ºС–Н больше, чем в этилене; sp-гибридизация затрудняет гомолитический разрыв связи ºС–Н с образованием радикалов, но облегчает гетеролитический разрыв этой связи с образованием ионов.
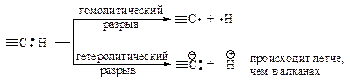
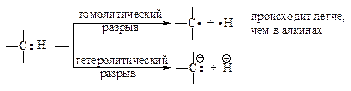
Физические свойства
Алкины малополярные соединения, их физические свойства сходны со свойствами алканов и алкенов: они также нерастворимы в воде, но растворимы в таких органических растворителях, как эфир, четырёххлористый углерод, бензол; плотность их меньше плотности воды. Низшие алкины - газы; температуры кипения алкинов повышаются с увеличением числа атомов углерода; разветвление цепи в молекуле изомера понижает температуру кипения.
Отличительным свойством ацетилена и его гомологов с одной алкильной группой является поглощение в области 3300 см-1 , соответствующее валентным колебаниям n (ºС–Н). Увеличение частоты в ряду n (Сsp3 –H), n (Сsp2 –H), n (Сsp –H) объясняется увеличением доли s-состояния гибридных орбиталей атомов углерода, образующих связи С–Н, при этом связь ºС–Н становится более короткой, прочной и более жёсткой.
Благодаря наличию частичных положительных зарядов на атомах водорода групп ºС–Н между атомом водорода ацетилена и атомами кислорода и азота других органических соединений (кетонов, эфиров, аминов) возникает сильное диполь-дипольное взаимодействие.
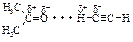
Поэтому ацетилен достаточно хорошо растворим в полярных органических растворителях.
Химические свойства
Химические свойства алкинов обусловлены их способностью реагировать двумя путями:
1). Посредством раскрытия кратной связи - реакции присоединения;
2). Посредством замещения атома водорода при углероде с тройной связью.
Реакции присоединеня
Галогенирование
Алкины, как и алкены, имеют доступные для атаки p-электроны и вступают в реакции электрофильного присоединения. Но из-за повышенной электроотрицательности sp-гибридизованных атомов углерода и укороченности связей поляризуемость p -электронов тройной связи СºС уменьшена, поэтому тройная углерод-углеродная связь по сравнению с двойной связью С=С менее реакционноспособна в отношении электрофильных реагентов.
Электрофильное присоединение галогенов (хлора, брома) к алкинам протекает аналогично реакции присоединения к алкенам.
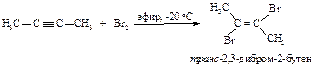
В первой быстройстадии p-электроны тройной связи отталкивают электроны ближайшего атома брома к удалённому атому брома; происходит поляризация молекулы брома и образование p-комплекса.
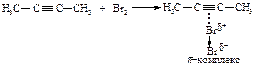
Во второй стадии p-комплекс перестраивается в циклический бромониевый катион, который является простейшим устойчивым сопряжённым соединением (число p-электронов равно двум). Эта стадия является медленной скорость лимитирующей.
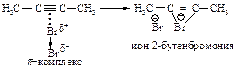
На третьей стадии происходит присоединение аниона брома преимущественно в транс-положение, со стороны, противоположной брому образовавшегося катиона, так как цис-присоединение стерически затруднено из-за большого объёма электронных оболочек брома.
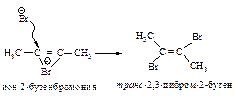
Эта реакция является примером реакции с высокой степенью стереоселективности.
При избытке брома в качестве конечного продукта бромирования ацетилена получается 2,2,3,3-тетрабромбутан.
Гидрогалогенирование
Алкины реагируют с галогеноводородами (хлороводородом, бромоводородом) подобно алкенам. Присоединение происходит в две стадии, по правилу Марковникова.
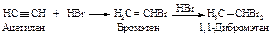
Механизм реакции. Образующийся в первой быстрой стадии p-комплекс во второй медленной стадии превращается в s -комплекс - карбокатион.
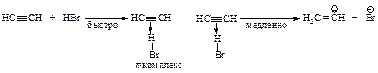
В третьей стадии происходит стабилизация карбокатиона – взаимодействие с анионом брома.
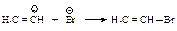
Взаимодействие бромалкена со второй молекулой бромоводорода происходит также по правилу Марковникова.
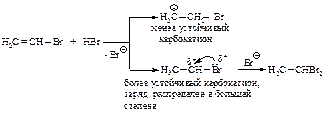
Гидратация
Присоединение воды к алкинам протекает в присутствии серной кислоты и солей двухвалентной ртути (реакция Кучерова).
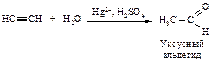
Гидратация гомологов ацетилена происходит в соответствии с правилом Марковникова.
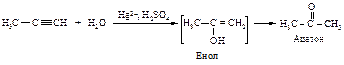
Ионы ртути образуют с молекулами алкинов p-комплексы, которые увеличивают растворимость алкинов в воде. Присоединение протона к алкину является стадией, определяющей скорость реакции. При этом разрывается p -связь, и образуется ненасыщенный спирт с группировкой, содержащей при углероде в sp2 -состоянии группу -ОН (енол).
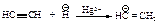
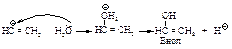
Енолы обычно неустойчивы и быстро превращаются в карбонильные соединения. В случае этенола, образующегося из ацетилена, конечным продуктом является ацетальдегид, из енолов, получающихся из других алкинов, образуются кетоны.
Механизм превращения енола I в альдегид состоит в следующем: протон присоединяется к углероду при двойной связи с образованием карбокатиона II, стабилизация которого происходит за счёт отрыва протона от кислорода с образованием уксусного альдегида.
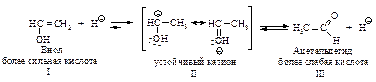
Перегруппировка енола в альдегид является примером превращения более сильной кислоты в более слабую. Протон легко отщепляется от кислорода енола I. Обратная реакция – отщепление протона от атома углерода с образованием енола I происходит очень трудно.
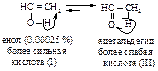
Равновесие между енолом и карбонилсодержащим соединением представляет собой прототропное равновесие, т.е. равновесие между структурами, которые отличаются положением водорода и кратной связи. Этот тип превращений называют также кето-енольной таутомерией (динамическая изомерия).
Каталитическая гидратация алкинов протекает легче, чем гидратация алкенов. Примером селективной гидратации тройной связи в присутствии двойной связи может служить реакция превращения винилацетилена в метилвинилкетон.
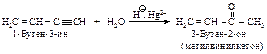
Гидроборирование
Другой способ присоединения элементов воды (Н и ОН) состоит в реакции присоединения борана по тройной связи с последующим окислением триалкенилборана пероксидом водорода в щелочной среде.
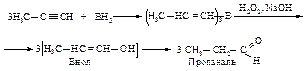
Образовавшийся виниловый спирт изомеризуется в более устойчивый альдегид (если алкин имеет концевую тройную связь) или кетон.
Нуклеофильное присоединение
В алкинах sp-гибридизованный атом углерода обладает более высокой электроотрицательностью, чем sp2-гибридизованный атом в алкенах и sp3-гибридизованный атом в алканах. Вместе с тем положительно заряженные ядра атомов углерода алкинов с внешней стороны экранированы в меньшей степени. Поэтому алкины вступают в реакции нуклеофильного присоединения AdN, на стадии определяющей скорость реакции, происходит присоединение нуклеофила.
Механизм реакции
Образование нуклеофила в реакции между спиртом и щелочью.
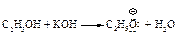
Первая стадия (медленная): нуклеофильное присоединение нуклеофила С2H5OӨ к алкину.
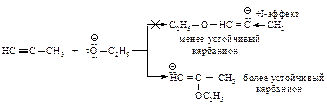
Вторая стадия (быстрая): стабилизация карбаниона путём отрыва протона от молекулы спирта, при этом регенерируется алкоксид-анион.
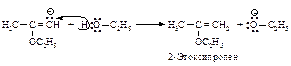
Синильная кислота так же присоединяется к ацетилену с образованием акрилонитрила. Реакция протекает в присутствии солей одновалентной меди. Стадией, лимитирующей скорость этой реакции, является нуклеофильное присоединение цианид-иона к тройной связи.
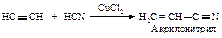
Кислотность алкинов
Водород группы ≡С–Н имеет небольшой положительный заряд, поэтому можно говорить о его ”кислотности“. Ацетилен как кислота реагирует с натрием в инертном растворителе (ксилол) с образованием соли – ацетиленида натрия.
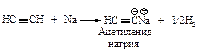
Ацетилен значительно уступает по кислотности воде и этиловому спирту, но превосходит по кислотности аммиак и углеводороды (таблица 10.3).
Таблица 10.3
Окисление алкинов
Окисление алкинов сильными окислителями (перманганат калия, концентрированная азотная кислота при нагревании) сопровождается расщеплением тройной связи с образованием карбоновых кислот. Эта реакция не играет значительной роли в органическом синтезе и используется для доказательства строения сложных природных соединений.
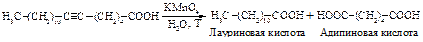
Окисление алкинов в нейтральной среде приводит к получению a-дикетонов.
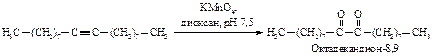
Тройная связь окисляется труднее, чем двойная. Благодаря этому можно осуществлять избирательное окисление енинов (соединений, содержащих двойную и тройную связи).
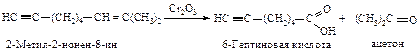
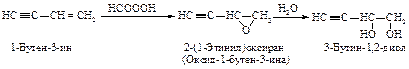
Озон является сильным окислителем и расщепляет тройную связь с образованием двух молекул карбоновых кислот.
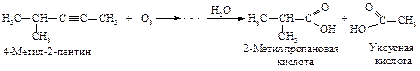
Способы получения
Простейший алкин - ацетилен - получают гидролизом карбида кальция, для образования которого из оксида кальция и углерода требуется большое количество тепла.
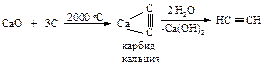
В промышленности ацетилен получают также окислительным пиролизом метана.
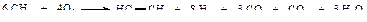
Ацетилен может служить исходным веществом для синтеза более сложных алкинов. На первой стадии образуется ацетиленид щелочного металла, который во второй стадии реагирует с первичным алкилгалогенидом.
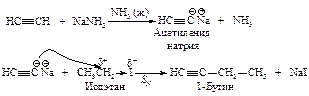
Таким же образом из моноалкилпроизводного синтезируют диалкилпроизводные (вводимые радикалы только первичные).
Другой метод синтеза алкинов основан на реакции соответствующих алкенов и галогенов с образованием вицинальных дигалогенидов, которые затем подвегают взаимодействию с амидом натрия в жидком аммиаке.
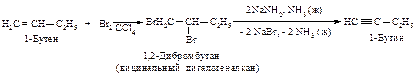
Применение спиртового раствора щелочи в реакции отщепления атомов галогена имеет следующий недостаток: в спиртовом растворе щелочи при повышенной температуре (>150 оС) происходит миграция тройной связи в средину углеводородной цепи.
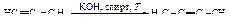
Алкины получают дегидрогалогенированием геминальных дигалогенидов, исходным соединением для получения последних могут служить кетоны и альдегиды.
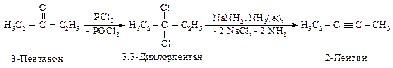
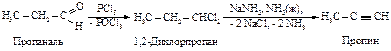
АРЕНЫ
К аренам относятся соединения, содержащие, по крайней мере, одну бензольную группировку*.
Исторически сложилось название этих соединений – ароматические. Появление его связано с тем, что на ранней стадии развития органической химии была выделена группа соединений, которые обладают приятным запахом или извлекались из душистых природных веществ. Родоначальником этих соединений оказался бензол. В современной химической литературе понятия "соединение ароматического ряда", "ароматический характер" означают сходство химических свойств соединений со свойствами бензола и не связаны с запахом этих соединений.
Строение бензола
Согласно теории резонанса, в любом случае, когда строение молекулы может быть изображено несколькими структурами, отличающимися только распределением электронов, ни одна из этих структур не адекватна рассматриваемой молекуле. Молекула представляет собой резонансный гибрид этих структур, называемых граничными. Каждая из граничных структур вносит свой вклад в гибрид: чем устойчивее структура, тем больше ее вклад.
Бензол представляет собой резонансный гибрид двух равноценных граничных структур: (I) и (II).
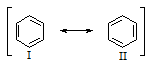
Изображение бензола в виде двух структур вовсе не предполагает их существование. Это означает, что строение бензола не может быть изображено ни структурой (I), ни структурой (II). Бензол имеет строение, промежуточное между ними. Поскольку граничные структуры (I) и (II) эквивалентны, их вклад в резонансную структуру одинаков. Бензол устойчивее, чем любая из граничных структур, на 154 кДж/моль.
Каждый атом углерода в молекуле бензола находится в sp2-гибридном состоянии и связан тремя s-связями с двумя атомами углерода и одним атомом водорода. Атомы углерода и водорода лежат в одной плоскости.
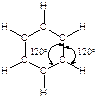
Четвертый валентный электрон атома углерода находится на 2р-орбитали, перпендикулярной плоскости молекулы. Эти 2р-орбитали состоят из двух одинаковых долей, одна из которых лежит выше, другая - ниже плоскости кольца; 2р-орбиталь каждого атома углерода перекрывается с 2р-орбиталями обоих соседних атомов углерода. В результате образуется замкнутая шести-p -электронная система в виде двух “бубликов”, один из которых лежит выше, а другой - ниже плоскости правильного шестиугольника.
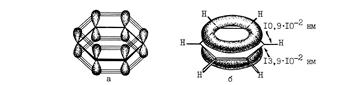
Рис. 11.1. Молекула бензола:
а - перекрывание 2р-орбиталей, образующих p -связи;
б - p-облака в виде двух "бубликов"
Благодаря коллективному взаимодействию всех шести p-электронов происходит выравнивание всех связей С–С по длине и кратности. Все связи углерод-углерод в молекуле бензола являются промежуточными между двойной и простой связями. Длина всех связей С–С в бензоле (13,9×10-2 нм) оказывается промежуточной между длиной двойной связи С=С в алкенах (13,4×10-2 нм) и расчётным значением длины простой связи С(sp2)–С(sp2), равным (14,8×10-2 нм).
Вторым следствием коллективного p-электронного взаимодействия является выигрыш энергии за счет резонанса структур (I) и (II), т.е. электронная и связанная с ней термодинамическая и кинетическая стабилизация бензола.
Ароматичность
Какие свойства должно проявлять вещество для того, чтобы его можно было отнести к ароматическим соединениям? Ароматичность можно определить с точки зрения кинетической устойчивости. Ароматическими называются соединения с молекулярной формулой, указывающей на высокую степень ненасыщенности, которые, однако, не реагируют как ненасыщенные соединения (кинетическая устойчивость), а вступают в реакции электрофильного замещения; для них характерна высокая термодинамическая устойчивость.
Условие ароматичности определяется правилом Хюккеля.
Ароматическими свойствами обладает соединение, если его строение удовлетворяет следующим требованиям:
Физические свойства
Физические свойства аренов похожи на свойства углеводородов других классов. Арены представляют собой малополярные соединения, плохо растворимые в воде, но растворимые в неполярных и слабополярных растворителях: гексане, эфире, четыреххлористом углероде.
Температура кипения аренов несколько выше, чем температура кипения углеводородов жирного ряда с тем же числом атомов углерода.
Температура кипения алкилбензолов растёт с увеличением молекулярной массы так же, как и в ряду алканов. Увеличение температуры кипения за счёт удлинения алкильной линейной цепочки на один атом углерода составляет 20-30 ºС. Разветвлённые алкилбензолы кипят при более низкой, а плавятся при более высокой температуре, чем их изомеры с линейной углеродной цепью.
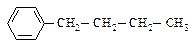
| 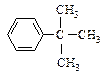
|
| н-Бутилбензол Ткип = 183 оС Тпл = -81 оС | трет-Бутилбензол Ткип = 169 оС Тпл = -58 оС |
Температуры кипения мета- и пара-изомеров диалкилбензолов приблизительно одинаковы, а температура кипения орто-изомера на несколько градусов выше. Плавление сопровождается разрушением межмолекулярных сил в кристалле; более симметричные молекулы более плотно упаковываются в кристаллической решетке. Чем прочнее кристаллическая решетка, тем выше температура плавления. Поэтому для дизамещенных производных бензола характерна такая закономерность: температура плавления наиболее симметричного пара-изомера обычно превышает температуры плавления менее симметричных орто- и мета-изомеров (см. табл. 11.2). Высокой степенью симметрии обладает бензол. Его температура плавления достаточно высока - плюс 5 оС.
Вследствие того, что растворение, так же как и плавление, связано с разрушением кристаллической решетки, пара-изомер менее растворим, чем орто- и мета-изомеры диалкилбензолов.
Для аренов характерны высокие значения показателя преломления (см. табл. 11.2). Это является следствием сопряжения p -электронов и повышенной поляризуемости сопряжённой p-электронной системы ароматического ядра. Особенно большое значение показателя преломления у стирола, так как в нем с p -электронами ароматического кольца дополнительно сопряжена винильная группа.
Таблица 11.2
Физические свойства аренов
| Название | Формула | Тпл, оС | Ткип, оС | Плотность | Показатель преломления |
| Бензол | 
| 5,5 | 80,0 | 0,879 | 1,5065 |
| Толуол | 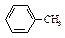
| -95,0 | 111,0 | 0,866 | 1,4961 |
| орто-Ксилол | 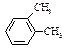
| -25,0 | 144,0 | 0,880 | 1,5055 |
| мета-Ксилол | 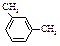
| -48,0 | 139,0 | 0,864 | 1,4972 |
| пара-Ксилол | 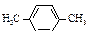
| 13,0 | 138,0 | 0,861 | 1,4958 |
| Этилбензол | 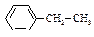
| -95,0 | 136,0 | 0,867 | 1,4959 |
| Стирол | 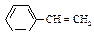
| -31,0 | 145,0 | 0,907 | 1,5462 |
| н-Пропилбензол | 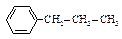
| -99,0 | 159,0 | 0,862 | 1,4920 |
| Изопропилбензол (кумол) | 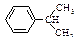
| -96,0 | 152,0 | 0,862 | 1,4915 |
Химические свойства
Для бензола характерны реакции замещения, в которых сохраняется устойчивая шести-p-электронная ароматическая система. Хотя p -электроны ароматического кольца в большей степени, чем p-электроны в алкенах, участвуют в связывании ядер углерода, они удерживаются все же слабее, чем s - электроны, и доступны для реагентов, любящих электроны - электрофилов. Следовательно, типичными реакциями аренов должны быть реакции электрофильного замещения ( SE Ar ).
Электрофильное замещение
Для аренов характерны следующие реакции электрофильного замещения
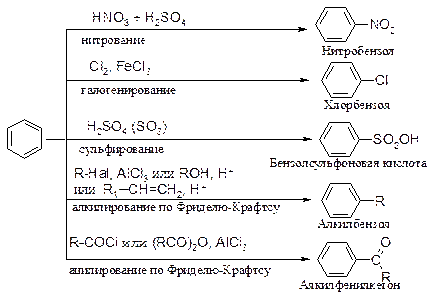
Нитрование
Азотная кислота очень медленно реагирует с бензолом. Для ускорения реакции нитрования бензола к азотной кислоте добавляют серную кислоту. Смесь концентрированных серной и азотной кислот называют нитрующей смесью.
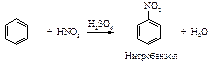
Образование электрофильной частицы - нитроний-катиона NO2Å происходит в реакции между азотной и серной кислотами. Вначале образуется протонированная азотная кислота.
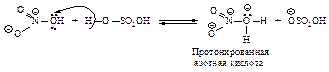
Эта реакция представляет собой кислотно-основное равновесие. Серная кислота является кислотой, а более слабая азотная кислота ведёт себя как основание, предоставляя пару электронов для образования связи с протоном. Протонированная азотная кислота распадается с образованием нитроний-катиона O2NÅ и молекулы воды.
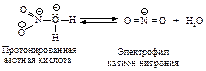
Первая стадия – медленная, положительно заряженный нитроний-катион – электрофильный реагент или электрофил - атакует доступное p - электронное облако бензола, вытягивая из него пару электронов. За счет этой пары электронов и происходит присоединение атома азота к атому углерода с образованием s - связи углерод-азот. Атакуемый атом углерода переходит из sp2- гибридного состояния в sp3- гибридное состояние. При этом возникает карбокатион, называемый также s-комплексом. Название “s -комплекс” указывает на образование s -связи углерода кольца с азотом электрофильного реагента.
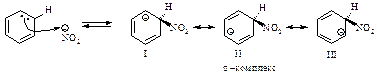
Каково строение s -комплекса? Оно не может быть показано структурой (I) с локализованными двойными связями и локализованным положительным зарядом. Строение s -комплекса должно быть изображено резонансным гибридом трёх граничных структур - (I), (II) и (III), которые отличаются только распределением электронов:
Четыре p - электрона распределены между пятью атомами углерода, находящимися в sp2- гибридном состоянии, положительный заряд не локализован на одном атоме углерода, а распределен между двумя орто- и одним пара-углеродными атомами относительно sp3- гибридизованного атома углерода. Распределение электронной плотности, а следовательно, и положительного заряда между несколькими атомами углерода ядра, делает карбокатион достаточно устойчивым. Энергия сопряжения этой системы незначительно отличается от энергии сопряжения бензола и составляет 109 кДж/моль. Именно благодаря такой стабилизации возможно образование карбокатиона из очень устойчивой молекулы бензола.
В дальнейшем строение s - комплекса будет изображаться структурой, в которой дуга обозначает четыре p-электрона, распределенные между пятью атомами углерода.
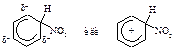
Вторая стадия – стабилизация карбокатиона. Первая стадия электрофильного замещения в ароматическом ядре подобна первой стадии электрофильного присоединения к алкенам. Но на второй быстрой стадии реакции происходит не присоединение нуклеофила, как в реакции электрофильного присоединения алкенов, а отщепление протона от sp3- гибридизованного атома углерода сопряженным анионом электрофила НOSO2O¯, два электрона вновь втягиваются в кольцо, атом углерода переходит из sp3-гибридного состояния в sp2-гибридное состояние с образованием более устойчивого шести p-электронного секстета.
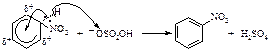
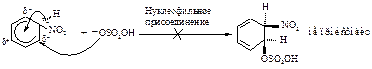
Это направление реакции имеет более низкую энергию активации (Е2< Е21), так как сопровождается образованием более устойчивого шести-p-электронного секстета.
Таким образом, электрофильное замещение представляет собой двухстадийный процесс, который представлен на диаграмме (рис. 11.2). Первая стадия (медленная) - присоединение электрофила к бензольному кольцу; вторая (быстрая) - отрыв протона сопряженным анионом электрофила.
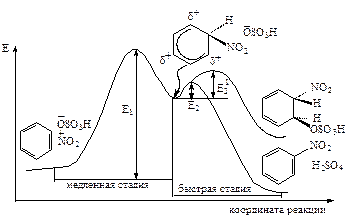
Рис.11.2. Энергетическая диаграмма реакции нитрования бензола.
Реакции галогенирования, сульфирования, алкилирования и ацилирования протекают по такому же механизму, отличие состоит только в способах образования электрофильной частицы.
Галогенирование.
Хлор и бром реагируют с бензолом в присутствии кислот Льюиса (AlCl3, FeCl3, FeBr3). На практике в качестве катализатора при галогенировании применяют, как правило, железные стружки. Катализатор в таком случае образуется непосредственно в реакционной массе при взаимодействии железных стружек с галогеном.
Электрофильная частица образуется в реакции галогена с катализатором. Эта реакция представляет собой кислотно-основное взаимодействие, продуктом которого является донорно-акцепторный комплекс (ДАК).
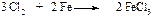
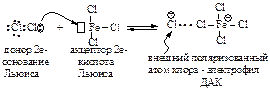
Внешний атом хлора становится достаточно электрононенасыщенным, чтобы атаковать бензольное ядро.
Сульфирование
Сульфирование бензола можно осуществить дымящей серной кислотой (Н2SО4+ SO3).
В этой реакции электрофильным реагентом является трёхокись серы SO3 - нейтральная молекула, в которой три электроотрицательных атома кислорода, связанные с атомом серы, делают последний электрононенасыщенным.
Стадия образования электрофильного реагента:
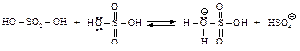
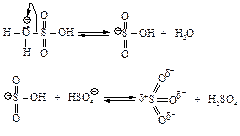
Алкилирование
Алкилирование по Фриделю-Крафтсу состоит в реакции бензола с алкилгалогенидами в присутствии кислот Льюиса (AlBr3, AlCl3, FeCl3, SbCl5, BF3, ZnCl2 и др.).
В реакции алкилирования по Фриделю-Крафтсу нельзя использовать в качестве алкилирующих агентов арилгалогениды Ar-Hal и винилгалогениды R-CH=CH-Hal.
Электрофильная частица образуется при взаимодействии алкилгалогенида (основание Льюиса) с катализатором (кислотой Льюиса). Взаимодействие протекает через следующие стадии.
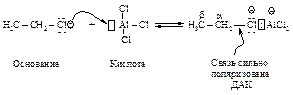
В более сложных первичных и вторичных галогеналканах первоначально образующийся карбокатион перегруппировывается в более устойчивый за счет миграции гидридиона или алкиланиона.
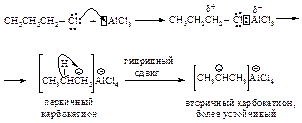
Если в качестве алкилирующих реагентов используются алкен или спирт, образование электрофилов протекает по схеме:
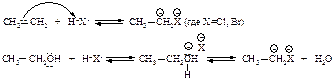
В более сложных алкенах и спиртах возможна перегруппировка так же, как и в случае галогеналканов.
Алкилирование по Фриделю-Крафтсу имеет ряд серьезных недостатков.
Во-первых, реакция алкилирования по Фриделю-Крафтсу связана с изомеризацией алкилирующего агента в ходе реакции, в результате чего образуется смесь изомерных продуктов алкилирования.
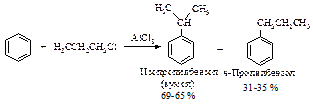
Основным продуктом алкилирования является изопропилбензол – результат взаимодействия бензола с вторичным карбокатионом (CH3)2CHÅ. Возможно, перегруппировка осуществляется на стадии образования ДАК.
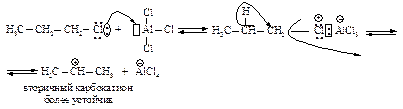
Для получения алкилбензолов с неразветвленной цепью атомов углерода используют двухстадийный синтез (п.11.7.4).
Во-вторых, образующийся при алкилировании бензола продукт является более реакционноспособным, чем бензол. Поэтому алкилирование аренов алкилгалогенидами при соотношении реагентов, близком к эквимолярному, приводит к образованию значительного количества продуктов полиалкилирования.
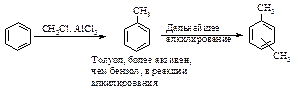
В этом отношении алкилирование сильно отличается от нитрования и галогенирования. Для того чтобы свести полиалкилирование к минимуму, используют большой избыток ароматического углеводорода. В этом случае он выполняет роль и реагента, и растворителя.
Еще одно ограничение для использования реакции алкилирования по Фриделю-Крафтсу связано с миграцией алкильной группы в конечном продукте (диспропорционирование продуктов). Например, алкилирование толуола хлористым изопропилом при 0 оС (AlCl3, растворитель – ацетонитрил CH3CN) приводит к смеси изомеров.
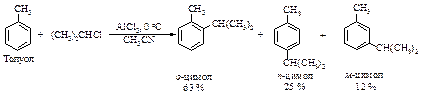
Однако при 25 оС в присутствии AlCl3 (2 моль) и HCl образуется только мета-изомер.
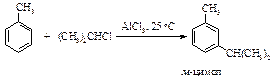
Из смеси трех изомерных цимолов в присутствии катализатора BF3 ×HCl уже через 10 минут образуется только мета-изомер.
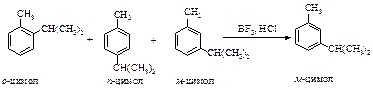
Алкилирование по Фриделю-Крафтсу относится к немногочисленной группе обратимых реакций электрофильного ароматического замещения, подчиняющихся термодинамическому контролю: в продуктах реакции, протекающей при более высокой температуре, преобладает наиболее термодинамически устойчивый 1,3-диалкил- или 1,3,5-триалкилбензол.
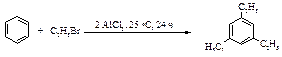
Изомеризация первоначально образующихся продуктов алкилирования происходит на стадии образования s-комплекса за счет 1,2-сдвига отрицательно заряженной алкильной группы (карбоаниона).
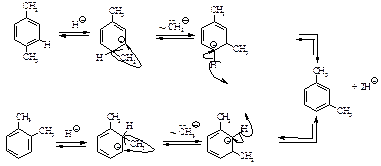
В результате перемещения группы CH3Θ в карбокатионах между тремя изомерами ксилола устанавливается равновесие, в котором всегда преобладает наиболее стабильный мета-изомер. В зависимости от температуры массовая доля м-ксилола в смеси равна 52-60 %, п-ксилола – 23-24 % и о-ксилола – 16-25 %. В более жестких условиях изомеризация алкилбензолов приобретает межмолекулярный характер. В результате из ксилолов образуется смесь, содержащая три-, тетра- и пентаметилбензолы наряду с толуолом и бензолом, происходит диспропорционирование.
Таким образом, при алкилировании правила ориентации соблюдаются только до тех пор, пока процесс протекает в мягких условиях, благоприятствующих кинетическому контролю (низкая температура, малые количества катализатора). Напротив, в условиях термодинамического контроля (более высокая температура, продолжительное время реакции, большие количества катализатора) алкилирование приводит преимущественно к мета-замещенным продуктам.
Для алкилирования ароматических углеводородов широко используется формальдегид. В присутствии минеральных кислот реакция бензола с формальдегидом приводит к дифенилметану с высоким выходом.
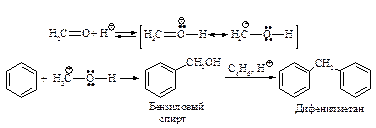
Ацилирование
Ацилирование - введение ацила R-C=O, или ароила Ar-C=O - происходит также в присутствии катализатора Фриделя-Крафтса При ацилировании ароматического кольца в качестве ацилирующего агента обычно используют хлорангидриды и ангидриды карбоновых кислот.
Активная частица – электрофил - образуется в реакции между хлорангидридом и хлоридом алюминия.
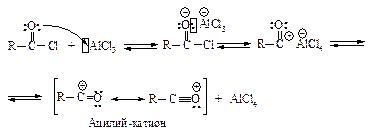
В случае использования в качестве ацилирующего агента ангидрида карбоновых кислот электрофил образуется в реакции между ангидридом и хлоридом алюминия.
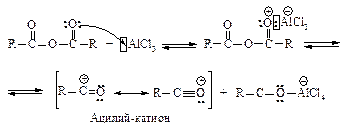
В реакцию ацилирования необходимо вводить эквимолекулярное количество катализатора, так как хлорид алюминия выводится из реакционной среды, давая устойчивое соединение с образующимся продуктом.
Ацилирование по Фриделю-Крафтсу лишено тех недостатков, которые присущи реакции алкилирования. При ацилировании вводится только одна ацильная группа, поскольку ароматические кетоны не вступают в дальнейшую реакцию ацилирования (так же, как и другие арены, содержащие сильные электроноакцепторные группы). Еще одним преимуществом этой реакции является отсутствие перегруппировок в ацилирующем агенте. Кроме того, для ацилирования не характерны реакции диспропорционирования продуктов.
В реакции алкилирования и ацилирования не вступают соединения, содержащие только электроноакцепторные группы (-NO2, -COOH, -CN). Ароматические кольца с группами (-NH2, -NHR, -NR2) не вступают в реакцию Фриделя-Крафтса из-за связывания кислоты Льюиса с основаниями.
Хлорметилирование
Хлорметильную группу можно ввести непосредственно в кольцо по способу, сходному с реакцией Фриделя - Крафтса, - взаимодействием бензола с формальдегидом и хлороводородом в присутствии хлористого алюминия или хлористого цинка.
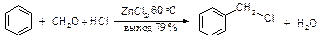
При хлорметилировании наблюдаются те же особенности, что и в реакции алкилирования по Фриделю-Крафтсу: такое же влияние ориентантов; кроме того, образуется некоторое количество полизамещённых продуктов. В приведённом примере получается в небольшом количестве п-дизамещённый продукт.
Формилирование
При помощи реакции, аналогичной синтезу кетонов по Фриделю-Крафтсу, можно ввести в бензольное кольцо формильную группу –СНО (реакция Гаттермана-Коха). Реакцию проводят в присутствии хлористого алюминия и однохлористой меди.
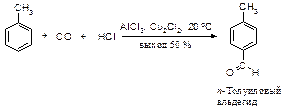
Возможно, что промежуточным неустойчивым продуктом этой реакции является формилхлорид, но как таковой он выделен не был.
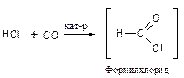
Ориентация и границы применения реакции Гаттермана-Коха приблизительно те же, что и при синтезе кетонов по Фриделю-Крафтсу, но выходы ниже. В обычных условиях этого метода бензол в реакцию не вступает и может быть использован в качестве растворитедя при формилировании других углеводородов.
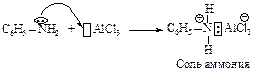
Реакции окисления
Реакции окисления в зависимости от условий и природы окислителя могут протекать по-разному. Молекулярный кислород при температуре около 100 оС окисляет изопропилбензол по радикальноцепному механизму.
Гидропероксид изопропилбензола можно разложить с образованием фенола и ацетона. Этот процесс разработали ученые Р.Ю. Удрис и П.Г. Сергеев (1942 г.).
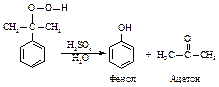
Окисление бензола молекулярным кислородом в присутствии пятиокиси ванадия приводит к образованию малеинового ангидрида.
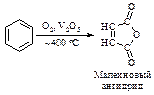
Озон действует на ароматическое ядро так же, как на углеводороды с двойными связями.
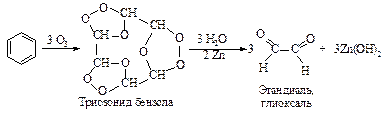
Сильные неорганические окислители (KMnO4, K2Cr2O7 + H2SO4) окисляют боковые цепи алкилароматических соединений до карбоксильных групп, связанных непосредственно с ароматическим кольцом.
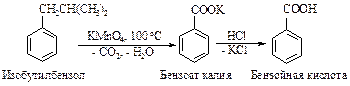
Трет-бутильная группа устойчива к окислению, так как не содержит водорода у a -углеродного атома.
Методы синтеза аренов
Бензол и многие его гомологи содержатся в нефти различного происхождения, в коксовых газах и каменноугольной смоле. В настоящее время в больших масштабах осуществляют дегидрирование, дегидроциклизацию, дегидроизомеризацию или высокотемпературный крекинг алифатических и алициклических углеводородов нефти до аренов.
Ароматизация алканов
Процесс превращения алканов в арены называется дегидроциклизацией, т.к. одновременно включает в себя две реакции: замыкание в цикл линейного углеводорода и отщепление водорода. Из гексана получается бензол, из гептана – толуол, из октана - смесь изомерных ксилолов. Другие арены этим способом не получают.
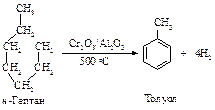
Для получения алкилбензолов существует ряд синтетических методов.
Реакция Вюрца – Фиттига
Аналогично получению алканов по реакции Вюрца алкилбензолы можно получить взаимодействием галогенбензола и галогеналкана с натрием. Наилучшие выходы получаются при использовании арилбромидов и первичных алкилбромидов.
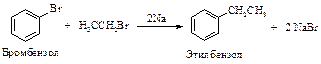
При этом в качестве побочных продуктов получаются бифенил и алкан, но они легко отделимы от алкиларенов.
Гомологи бензола могут быть получены также при взаимодействии фенилмагнийгалогенидов с галогеналканами.
Алкилирование бензола
При действии на бензол алкилирующих агентов (галогеналканов, алкенов, спиртов) в присутствии кислот Льюиса, а также сильных кислот образуется алкилбензол.
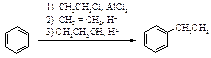
11.7.4.Ацилирование с последующим восстановлением
Реакция ацилирования может быть использована в двухстадийном синтезе алкилбензолов с неразветвлённой цепью атомов углерода: первая стадия - ацилирование, вторая - восстановление полученного кетона амальгамированным цинком в концентрированной соляной кислоте (восстановление по Клеменсену).
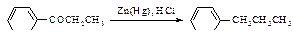
Нафталин
Нафталин - двухядерный конденсированный арен.
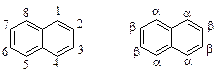
В молекуле нафталина все атомы лежат в одной плоскости, каждый атом углерода находится в состоянии sp2-гибридизации и связан с соседними атомами углерода и водорода s-связями. В результате перекрывания p-орбиталей образуется p-электронное облако, лежащее над и под плоскостью колец. Число сопряженных p - электронов соответствует правилу Хюккеля N = 4n + 2, где n = 2 и N = 10.
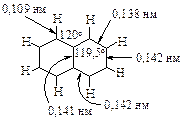
Строение молекулы нафталина можно представить как резонансный гибрид трех структур I, II, III.
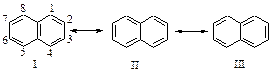
Рассмотрение этих структур показывает, что связи С1 - С2, С3 - С4 , С5 - С6 , С7 - С8 являются двойными в двух структурах и простыми в одной структуре. Связи С2 - С3 и С6 - С7 - простые в двух структурах и двойные - только в одной. Ввиду неполного выравнивания связей энергия сопряжения нафталина меньше, чем энергии сопряжения двух молекул бензола 306 кДж/моль, и равняется 255 кДж/моль.
Нафталин как ароматическое соединение устойчив в реакциях присоединения, характерных для ненасыщенных веществ, и, напротив, подобно бензолу склонен к реакциям электрофильного замещения, в которых сохраняется устойчивая p - электронная система.
Реакции электрофильного замещения в нафталине происходят легче, чем в бензоле, так как в медленной стадии электрофильного замещения в бензоле разрушается сопряжение в 153 кДж/моль, в то время, как при образовании s-комплекса из нафталина потеря энергии сопряжения равняется разности энергий сопряжения нафталина 255 кДж/моль и бензола 153 кДж/моль, то есть составляет 102 кДж/моль. Хлорирование и бромирование происходят настолько легко, что в них не используют катализатор.
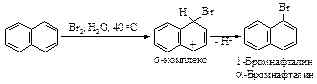
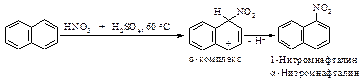
Нитрование и галогенирование нафталина происходят исключительно в a-положение, так как при этом в медленной стадии образуется s -комплекс более устойчивый, чем s -комплекс получающийся при атаке в b- положение: a - s -комплекс может быть изображен с помощью двух граничных структур (I) и (II), в которых сохраняется ароматический секстет, в то время как b - s -комплекс представлен лишь одной граничной структурой с сохранившимся ароматическим секстетом другого кольца.
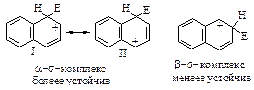
Сульфирование нафталина протекает по двум направлениям. В условиях кинетического контроля (невысокая температура, сравнительно короткое время протекания реакции) образуется преимущественно 1(a)–нафталинсульфоновая кислота. Однако ввиду обратимости реакции ароматического сульфирования и большей термодинамической устойчивости 2(b)–нафталинсульфоновой кислоты по сравнению с 1(a)–нафталинсульфоновой кислотой при повышенной температуре и достаточно продолжительном протекании реакции (условия термодинамического контроля) будет накапливаться наиболее устойчивый, но медленнее образующийся продукт - 2-нафталинсульфоновая кислота.
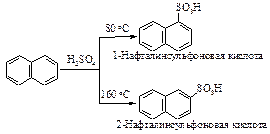
Ориентация в реакциях электрофильного замещения в производных нафталина происходит в соответствии со следующими правилами:
а). Электронодонорная группа, активирующая электрофильное замещение, направляет электрофильный реагент в то кольцо, в котором она находится. Если эта группа находится в положении 1, электрофильный реагент замещает водород в положение 2 или положении 4; электронодонорный ориентант в положении 2 направляет электрофил в положение 1.
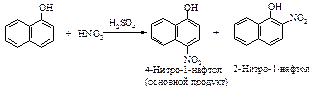
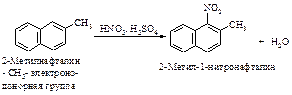
Такое направление замещения можно объяснить следующим образом. Ориентант оказывает наибольшее влияние на то кольцо, с которым он связан. Поэтому наиболее успешной является атака электрофилом EÅ кольца с электронодонорной группой G, в котором может лучше распределиться положительный заряд.
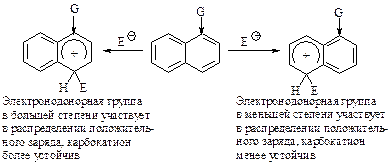
б). Электроноакцепторная группа направляет электрофильный реагент в другое незамещенное кольцо (в положение 5 или 8 при галогенировании и нитровании).
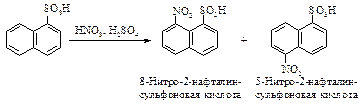
Восстановление нафталина в отличие от бензола можно провести химическими восстановителями. Первая стадия протекает достаточно легко, вторая - восстановление бензольного кольца в тетралине – протекает в жестких условиях, сходных с условиями гидрирования бензола.
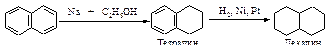
Окисление нафталина кислородом в присутствии пятиокиси ванадия приводит к образованию фталевого ангидрида.
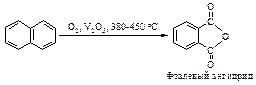
Нафталин окисляется смесью K2Cr2O7 и H2SO4 до фталевой кислоты.
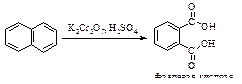
Если в одном из колец есть заместитель, то окисляется кольцо с повышенной электронной плотностью.

Антрацен и фенантрен
Антрацен и фенантрен являются ароматическими соединениями. Они представляют собой плоские циклические структуры, содержащие замкнутое p -электронное облако, расположенное ниже и выше плоскости колец. Число p -электронов в соответствии с правилом Хюккеля равно 4n + 2 = 4 × 3 + 2 = 14.
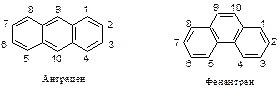
Антрацен можно рассматривать как резонансный гибрид структур I-IV.

Его энергия резонанса составляет 352 кДж/моль.
Фенантрен можно представить резонансным гибридом структур V-IX.
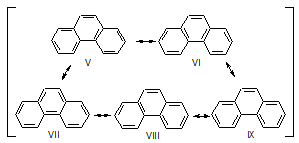
Резонансная энергия фенантрена 386 кДж/моль.
Антрацен и фенантрен вступают в реакции электрофильного замещения. Их активные положения 9 и 10 находятся в среднем кольце, так как при атаке в эти положения сохраняется ароматичность двух боковых бензольных систем с энергией резонанса 153×2=306 кДж/моль. При атаке в боковые кольца сохраняется ароматичность одного нафталинового фрагмента с энергией резонанса 256 кДж/моль.
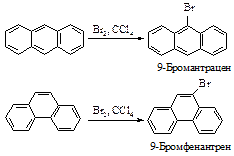
Вывод об активности положений 9 и 10 справедлив как для электрофильного замещения, так и для реакций окисления и восстановления.
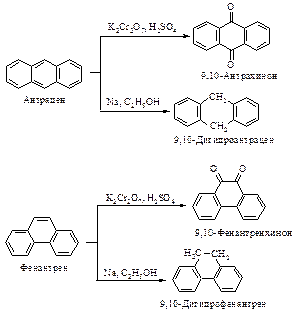
Пятичленные гетероциклы
Строение
Простейшие пятичленные гетероциклы: пиррол, фуран, тиофен - содержат один гетероатом.
В соответствии с представленными формулами каждое из этих соединений будет обладать свойствами сопряженного диена и соответственно - свойствами амина R-NH-R, простого эфира R-O-R и тиоэфира (сульфида) R-S-R. Однако пиррол не обладает основными свойствами, типичными для аминов, тиофен не вступает в реакции окисления, типичные для сульфидов. Для них характерна способность вступать в реакции электрофильного замещения: нитрование, сульфирование, галогенирование, ацилирование по Фриделю-Крафтсу. Числовые значения теплот сгорания указывают на наличие значительной энергии резонансной стабилизации - 67-117 кДж/моль. Это несколько меньше, чем энергия резонанса бензола (153 кДж/моль), но гораздо больше, чем аналогичная величина для большинства сопряженных диенов (13 кДж/моль). Следовательно, эти соединения являются ароматическими.
Рассмотрим строение пятичленных гетероциклов на примере пиррола. Четыре атома углерода и атом азота находятся в sp2-гибридном состоянии и затрачивают три гибридные орбитали на образование двух s-связей с другими атомами кольца и одним атомом водорода. У каждого атома углерода остается один электрон, а у атома азота - два на р-орбитали. При p-перекрывании р-орбиталей образуются p-облака выше и ниже плоскости кольца, содержащие шесть электронов, - ароматический секстет.
| 
|
Структура пиррола может быть представлена резонансным гибридом структур I-V.
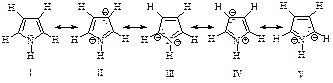
Сравнение дипольных моментов пирролидина 5,18×10-30 Кл×м (1,57Д) и пиррола 5,94×10-30 Кл×м (1,8Д) показывает, что структуры II-V вносят значительный вклад в резонансный гибрид.
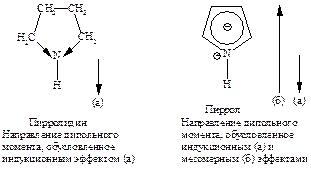
Данные, полученные при измерении длин связей в молекуле пиррола, дипольных моментов, УФ-спектров, теплот сгорания, подтверждают наличие делокализованной p-электронной системы.
Следствием делокализации четырех p-электронов атомов углерода и двух электронов гетероатома является склонность к реакциям электрофильного замещения, в которых сохраняется p-электронная система.
В отличие от вторичных алифатических аминов, для которых КВ»10-3-10-4, пиррол - более слабое основание (КВ =2,5×10-14). Это объясняется тем, что свободная пара электронов азота, которая обусловливает основные свойства азотсодержащих соединений, вовлечена в p-электронное облако и не может быть предоставлена для образования связи с протоном.
Высокая электронная плотность в кольце пиррола обусловливает высокую реакционную способность пиррола в реакциях электрофильного замещения.
Структуры фурана и тиофена аналогичны структуре пиррола. Атом кислорода в фуране и атом серы в тиофене подают два электрона в p-электронное облако и ведут себя как высокореакционноспособные соединения, подобные бензолу.
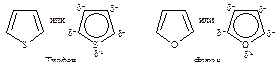
Сравнивая электроотрицательность серы, азота и кислорода, можно ожидать, что вклад структур, подобных II - V, будет наиболее значительным для тиофена и наименее - для фурана, содержащего наиболее электроотрицательный элемент - кислород. Действительно, ароматический характер усиливается при переходе от фурана к пирролу и далее к тиофену, энергии резонанса составляют соответственно 67·103, 88·103, 117·103 Дж/моль. Фуран, наименее ароматичный из этих трех соединений, вступает в реакции диенового синтеза в качестве диена.
14 .1.2. Химические свойства
Пиррол, фуран и тиофен, как и другие ароматические соединения, вступают в реакции электрофильного замещения: нитрование, сульфирование, галогенирование, ацилирование по Фриделю-Крафтсу.
Пиррол и фуран более реакционноспособные соединения, чем бензол, и сходны с наиболее активными производными бензола: аминами и фенолами. Тиофен менее реакционноспособен, чем пиррол и фуран, но более активен, чем бензол.
Электрофильное замещение происходит в положение 2.
В реакции электрофильного замещения лимитирующей стадией является стадия образования s -комплекса в результате присоединения электрофильного реагента к атому углерода кольца.
Реакции электрофильного замещения в пирроле представлены на рис. 14.1.
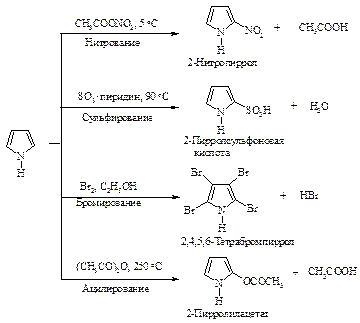
Рис. 14.1. Реакции электрофильного замещения в пирроле
Нитрование и сульфирование пиррола из-за чувствительности к протонным кислотам (это свойство называется ацидофобностью) проводят в отсутствие протонных кислот. Реакция бромирования и ацилирования по Фриделю-Крафту протекает без участия катализатора.
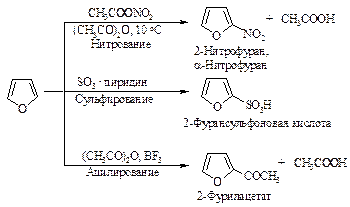
Фуран в реакциях электрофильного замещения напоминает пиррол. Как и пиррол, он является ацидофобным соединением: в присутствии протонных кислот кольцо фурана раскрывается. Хлорирование и бромирование фурана протекает очень бурно и с трудом поддается контролю. Ацилирование требует применения в качестве катализатора мягкой кислоты Льюиса.
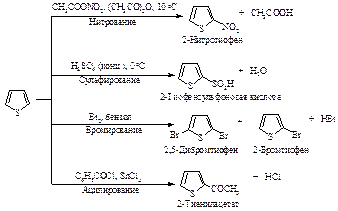
Тиофен менее реакционноспособен по сравнению с фураном и пирролом, он может сульфироваться в условиях высокой кислотности. Бромирование может проводиться направленно с образованием 2-бром- и 2,4-дибромтиофена.
Способы получения
Некоторые замещенные фурана, тиофена и пиррола могут быть получены из ациклических соединений через реакцию циклизации, например:
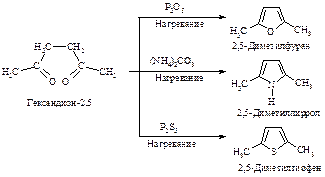
Фуран, тиофен, пиррол могут взаимно превращаться друг в друга (Ю.К. Юрьев) в токе Н2О, Н2S и NH3 соответственно при температуре 400-500 оС в присутствии Al2O3.
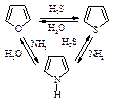
Пиридин
Пиридин является шестичленным ароматическим гетероциклом.
Строение пиридина
В пиридине атом азота, как и все атомы углерода, находится в sp2-cостоянии. Каждый из пяти атомов углерода и атом азота связаны с соседними атомами кольца с помощью двух sp2-гибридизованных орбиталей, третья sp2-гибридизованная орбиталь атома углерода затрачивается на образование связи с атомом водорода, а на третьей sp2-гибридизованной орбитали атома азота находится пара электронов. Пять негибридизованных р-облаков атомов углерода и одно р-облако атома азота, перекрываясь, образуют единую p -электронную систему: два облака - над и под плоскостью кольца.
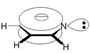
В плоском шестиугольнике пиридина все углерод-углеродные связи имеют одинаковую длину (13,9·10-2 нм), промежуточную между длиной простой Csp 2–Csp 2 (14,8·10-2 нм) и двойной C=C связей (13,4·10-2 нм). Обе связи азот-углерод также имеют одинаковую длину (13,7·10-2 нм), меньшую, чем длина простой связи С-N (14,7·10-2 нм), но большую, чем длина двойной связи С=N (12,8·10-2 нм).
Делокализация шести р-электронов кольца пиридина обусловливает значительную устойчивость p -электронного облака. Действительно, теплота сгорания пиридина указывает на существенную энергию резонанса 96 кДж/моль.
Строение пиридина можно изобразить резонансным гибридом граничных структур I-V.
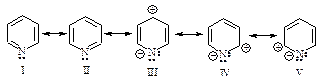
Биполярные структуры III-V вносят значительный вклад в резонансный гибрид, так как дипольный момент пиридина, равный 7,45×10-30 Кл×м (2,26 Д), значительно больше, чем дипольный момент пиперидина 3,86×10-30 Кл×м (1,17 Д).
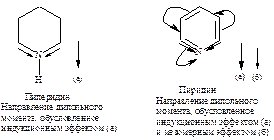
Для пиридина, как для ароматического соединения, характерны реакции электрофильного замещения, в которых сохраняется устойчивая p -электронная система.
Химические свойства
В реакциях электрофильного замещения пиридин ведет себя как сильно дезактивированное производное бензола, подобно нитробензолу. Он нитруется, сульфируется и галогенируется только в очень жестких условиях (более жестких, чем нитробензол). Замещение происходит в положение 3.
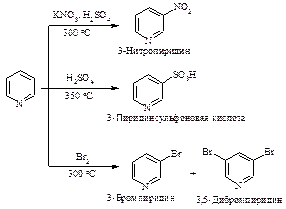
Низкая реакционная способность пиридина, как и нитробензола, объясняется тем, что образующиеся при атаке электрофилом любого положения кольца карбокатионы менее устойчивы, чем карбокатион, возникающий при атаке бензольного ядра, из-за присутствия в кольце электроотрицательного азота. Кроме того, в кислой среде кольцо еще более дезактивируется из-за образования иона пиридиния C6H5NHÅ.
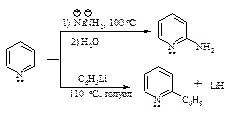
Реакционная способность пиридина в реакциях нуклеофильного замещения настолько велика, что замещению подвергается даже гидрид-ион НӨ. Примером нуклеофильного замещения в пиридине являются реакции Чичибабина: аминирование амидом натрия и арилирование (или алкилирование) с помощью литийорганических соединений.
Атака нуклеофила протекает преимущественно в положение 2 и 4, так как при этом образуется более устойчивый анион: отрицательный заряд частично несет электроотрицательный азот; s-комплекс, возникающий при атаке в положение 3, менее устойчив: ни в одной из граничных структур отрицательный заряд не принадлежит азоту.
Все эти s-комплексы более устойчивы, чем анион, через образование которого происходит нуклеофильное замещение в бензоле. Это обусловлено присутствием в ядре пиридина электроотрицательного азота.
Таким образом, одна и та же причина определяет более низкую реакционную способность пиридина в реакциях электрофильного замещения и более высокую реакционную способность его в реакциях нуклеофильного замещения по сравнению с бензолом.
В пиридине на атоме азота имеется неподеленная пара электронов, которая может обобществляться с протоном. Пиридин является более слабым основанием (КВ=2,3·10-9), чем алифатические амины R1R2NH (КВ~ 10-4). Для объяснения такой зависимости основности от строения можно провести аналогию между электроотрицательностью атома углерода в различных гибридных состояниях и электроотрицательностью атома азота в sp3- и sp2- состояниях:
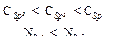
¾¾¾¾¾¾¾®
Электроотрицательность увеличивается
Пара электронов, обусловливающая основность пиридина, занимает sp2-орбиталь атома азота, она находится ближе к ядру. Электроны удерживаются ядром сильнее и менее доступны для обобществления с протоном, чем пара электронов sp3-гибридизованного атома азота.
Благодаря наличию пары электронов на атоме азота пиридин является нуклеофилом и реагирует с галогеналканами с образованием четвертичных солей.
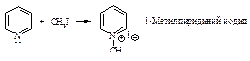
Восстановление. При каталитическом гидрировании пиридина образуется пиперидин С5Н11N.
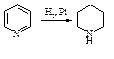
Хинолин
Хинолин представляет собой конденсированную систему, содержащую бензольное и пиридиновое кольца.
Свойства хинолина соответствуют свойствам нафталина, содержащего электроноакцепторную группу в положении 1.
Хинолин вступает в реакции электрофильного замещения. При взаимодействии со смесью концентрированных азотной и серной кислот происходит нитрование в положения 5 и 8.
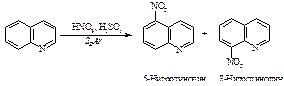
При обработке хинолина дымящей серной кислотой образуются сульфоновые кислоты.
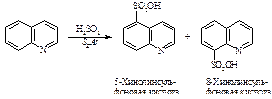
Хинолин так же, как и пиридин, вступает в реакции нуклеофильногозамещения: взаимодействует с амидом натрия и фениллитием.
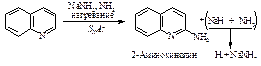
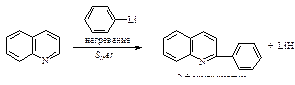
При окислении перманганатом калия образуется дикарбоновая кислота.
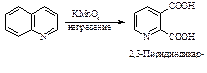
Наиболее удобным путем получения хинолина является метод Скраупа: взаимодействие анилина с глицерином и нитробензолом в присутствии серной кислоты и сульфата железа (II).
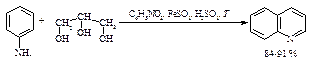
Синтез состоит из следующих стадий.
1). Дегидратация глицерина под действием концентрированной серной кислоты.
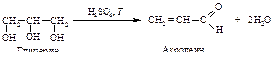
2). Нуклеофильное присоединение анилина к акролеину.

3). Электрофильная атака ароматического кольца электронодефицитным углеродом с последующей дегидратацией образовавшегося спирта.
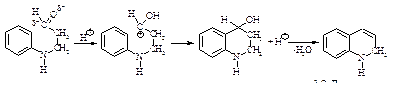
4). Окисление нитробензолом.
Сульфат железа FeSO4 сдерживает бурное течение реакции.
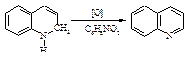
ГАЛОГЕНАЛКАНЫ
Галогеналканы имеют общую формулу СnH2n-1Х, где X - F, Cl, Br, I. Вследствие высокой электроотрицательности галогена связь галоген - углерод является сильно полярной Сd+®Хd-. Галоген, связанный с электронодефицитным углеродом, можно легко заменить на частицу, богатую электронами, - реагент - нуклеофил Nu («ядро любящий»). Нуклеофильными реагентами являются отрицательно заряженные ионы и нейтральные молекулы, имеющие свободную пару электронов.
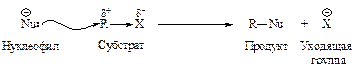
Такие реакции называются реакциями нуклеофильного замещения - SN.
Другой характерный тип превращений галогеналканов - реакции отщепления (элиминирования Е) под действием основания с образованием алкенов. Под влиянием электроотрицательного галогена связанный с ним атом углерода (Сa) приобретает некоторый положительный заряд, на соседнем атоме (Сb) также возникает заряд, но меньший. Тем не менее под влиянием частично заряженного b-углерода находящийся около него водород становится кислым и под действием сильного основания может отщепиться в виде протона (d+>d'+).
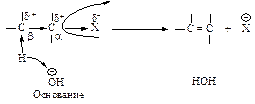
Таким образом, для галогеналканов характерны два главных типа превращений: нуклеофильное замещение и элиминирование. Кроме того, галогеналканы дают магнийорганические соединения R-MgX, важные в синтетическом отношении.
Нуклеофильное замещение
Благодаря доступности галогеналканов и легкости, с которой они вступают в реакции, круг этих реакций очень широк. Наиболее важные из них приведены в таблице 15.1.
Метилгалогениды CH3-X, первичные RCH2-X, вторичные R1R2CH-X, третичные R1R2R3-X алкилгалогениды взаимодействуют с нуклеофильными реагентами по разным механизмам в зависимости от строения алкила.
Таблица 15.1-Реакции нуклеофильного замещения
| Нуклеофил Nu | Продукт реакции R-Nu |
| НО– или Н2О | Спирт ROH |
| R1O- или R1OH | Простой эфир ROR1 |
| 
|
| NºC- | Нитрил карбоновой кислоты R-CºN |
| NO2- | Нитросоединение R- NO2 и/или R-ONO |
| NH2– | Амин R-NH2 |
| NH3 | Соль первичного амина RNH3+X- |
| R1NH2, R1R2NH | Соль вторичного или третичного амина RR1NH2+X-, RR1R2NH+ X- |
| R1R2R3NH | Четвертичная аммониевая соль (ЧАС) RR1R2R3N+ X- |
| R1CºC- | Алкины R1CºC-R |
| R1C- | R1C-R (реакция Вюрца) |
| I- | Иодиды R-I |
| SH-, R1–S-, -S–S- | Тиолы RSH, сульфиды R1SR, дисульфиды RSSR |
Галогенирование алканов
ГАЛОГЕНАРЕНЫ
Галогенаренами называются соединения, содержащие атом галогена, связанный непосредственно с ароматическим кольцом.
Синтез спиртов и кислот
Магнийорганические соединения присоединяются к карбонильным соединениям по двойной связи углерод - кислород.
Для синтеза первичных спиртов используют в качестве карбонильного соединения муравьиный альдегид.
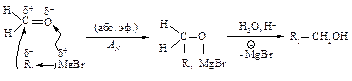
Для получения вторичных спиртов в качестве карбонильного соединения используют соответствующий альдегид.
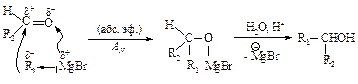
В случае синтеза третичных спиртов исходное карбонильное соединение - кетон.
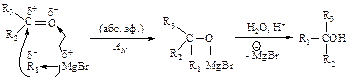
Для получения первичных спиртов, содержащих на два атома углерода больше, чем в магнийорганическом исходном соединении, используют окcид этилена.
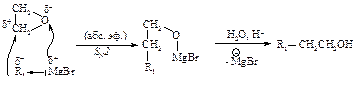
Как выбрать карбонильное соединение для синтеза определенного спирта? Предположим, необходимо получить 2-фенил-2-бутанол. Мысленно расщепляем молекулу спирта около углерода, несущего спиртовую группу. Та часть молекулы, которая содержит кислород, входила в исходное карбонильное соединение, другая - в реактив Гриньяра.
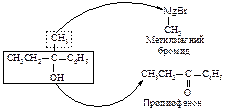
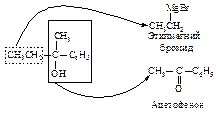
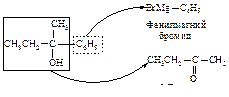
Таким образом, для синтеза указанного спирта можно выбрать три пары соединений.
При взаимодействии реактива Гриньяра с углекислым газом происходит присоединение его по двойной связи, как и в реакции с карбонильными соединениями, в результате образуется соль, из которой карбоновую кислоту выделяют действием разбавленного раствора минеральной кислоты.
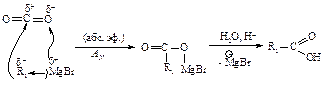
Библиографический список
1.Моррисон, Р. Органическая химия. – М. : Мир, 1974. – 1132 с.
2.Терней, А. Современная органическая химия : 2 т. – М. : Мир, 1991. – Т. 1. – 670 с.; Т. 2. – 615 с.
3.Робертс, Дж. Основы органической химии : в 2 т. / Дж. Моррисон, М. Кассерио. – 2-е изд. – М. : Мир, 1978. – Т. 1. – 842 с.; Т. 2. – 888 с.
4.Шабаров, Ю. С. Органическая химия : в 2 кн. – М. : Химия, 1994. – 848 с.
5.Травень, В. Ф. Органическая химия : 2 т. – М. : ИКЦ Академкнига, 2004. – Т. 1. – 727 с.; Т. 2. – 582 с.
ОГЛАВЛЕНИЕ
| Введение …………………………………………………... | 3 |
| 1. Классификация органических соединений ……………….. | 4 |
| 2. Классификация органических реакций ……………………. | 5 |
| 3. Способы образования ковалентной связи ……………….. | 7 |
| 4. Гибридизация атомных орбиталей и форма органических молекул ……………………………………………………….. | 8 |
| 4.1. sp 3-Гибридизация ………………………………………….. | 9 |
| 4.2. s р2-Гибридизация …………………………………………... | 10 |
| 4.3. sp-Гибридизация …………………………………………… | 11 |
| 5.АЛКАНЫ …………………………………………………….. | 12 |
| 5.1. Физические свойства ……………………………………... | 15 |
| 5.2. Химические свойства ………………………………………. | 16 |
| 5.3. Методы синтеза алканов …………………………………. | 26 |
| 6. СТЕРЕОИЗОМЕРИЯ. ЭНАНТИОМЕРИЯ ……………….. | 29 |
| 6.1. Энантиомеры. Хиральность. Условия хиральности ………. | 29 |
| 6.2. Плоскополяризованный свет. Оптическая активность … | 31 |
| 6.3. Строение молекул и оптическая активность ……………. | 31 |
| 6.4. Обозначение конфигураций ……………………………… | 31 |
| 7. ЦИКЛОАЛКАНЫ …………………………………………... | 35 |
| 7.1 Номенклатура. Изомерия …………………………………. | 35 |
| 7.2. Физические свойства ……………………………………... | 36 |
| 7.3. Типы напряжения ………………………………………… | 37 |
| 7.4. Строение …………………………………………………... | 37 |
| 7.5. Химические свойства …………………………………….. | 43 |
| 7.6. Способы получения ………………………………………. | 45 |
| 8. Алкены ……………………………………………………. | 46 |
| 8.1. Физические свойства ……………………………………... | 47 |
| 8.2. Химические свойства …………………………………….. | 48 |
| 8.3. Способы получения алкенов …………………………….. | 73 |
| 9. ДИЕНЫ ……………………………………………….. | 75 |
| 9.1. Устойчивость сопряженных диенов …………………….. | 76 |
| 9.2. Химические свойства …………………………………….. | 78 |
| 9.3. Способы получения ………………………………………. | 87 |
| 10. АЛКИНЫ …………………………………………….. | 88 |
| 10.1. Физические свойства ……………………………………. | 90 |
| 10.2. Химические свойства …………………………………… | 91 |
| 10.3. Способы получения ……………………………………... | 102 |
| 11. АРЕНЫ ……………………………………………… | 103 |
| 11.1. Сравнение свойств бензола со свойствами алкенов ….. | 103 |
| 11.2. Теплота гидрирования. Энергия резонанса …………… | 104 |
| 11.3. Строение бензола ………………………………………... | 105 |
| 11.4. Ароматичность …………………………………………... | 107 |
| 11.5. Физические свойства ……………………………………. | 107 |
| 11.6. Химические свойства …………………………………… | 109 |
| 11.7. Методы синтеза аренов …………………………………. | 123 |
| 12. Электрофильное замещение в производных бензола ……………………………………………………….. | 125 |
| 12.1. Влияние заместителей на реакционную способность бензольного кольца …………………………………….. | 127 |
| 12.2. Влияние заместителя на выбор места электрофильной атаки ……………………………………………………… | 128 |
| 13. Многоядерные ароматические соединения ……………… | 134 |
| 13.1. Нафталин …………………………………………………. | 134 |
| 13.2. Антрацен и фенантрен …………………………………... | 138 |
| 14. Гетероциклические соединения ……………… | 140 |
| 14.1. Пятичленные гетероциклы ……………………………… | 141 |
| 14.2. Пиридин ………………………………………………….. | 146 |
| 14.3. Хинолин ………………………………………………….. | 150 |
| 15. ГАЛОГЕНАЛКАНЫ ………………………………………. | 152 |
| 15.1. Нуклеофильное замещение ……………………………... | 153 |
| 15.2. Реакции отщепления (элиминирование) ……………….. | 166 |
| 15.3. Методы синтеза галогеналканов ……………………….. | 171 |
| 16. ГАЛОГЕНАРЕНЫ ………………………………………… | 172 |
| 16.1. Причина низкой реакционной способности галогенаренов . | 172 |
| 16.2. Нуклеофильное замещение, протекающее через стадию образования дегидробензола, - отщепление – присоединение ……………….. | 173 |
| 16.3. Бимолекулярное нуклеофильное замещение SN2Ar …… | 174 |
| 16.4. Ориентация при нуклеофильном замещении в ароматическом кольце ………………………………………… | 176 |
| 17. МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ …………... | 177 |
| 17.1. Получение и строение магнийорганических соединений …… | 177 |
| 17.2. Синтез спиртов и кислот ………………………………... | 178 |
| Библиографический список ………………………... | 180 |
Учебное издание
Органическая химия
Часть 1
УГЛЕВОДОРОДЫ.
ГАЛОГЕНПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Составители: Бетнев Александр Федорович
Колпащикова Ия Семеновна
Кофанов Евгений Романович
Алов Евгений Михайлович
Редактор Л.С. Кокина
План 2007
Подписано в печать 10.07.2012. Формат 60х84 1/16. Бумага белая.
Печать ризограф. Усл. печ. л.14,25 Уч.-изд. л. Тираж 350. Заказ 1714
Ярославский государственный технический университет
150023, Ярославль, Московский пр., 88
Типография Ярославского государственного технического университета
150000, Ярославль, ул. Советская, 14а
Органическая химия
Часть 1
УГЛЕВОДОРОДЫ.
Ярославль 2012
* По Льюису, основание - соединение, предоставляющее пару электронов для образования связи, донор пары электронов; кислота - акцептор пары электронов.
* В литературе употребляются такие изображения этой группировки:
Органическая химия
Часть 1
УГЛЕВОДОРОДЫ.
ГАЛОГЕНПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Рекомендовано
научно-методическим советом
университета в качестве учебного пособия
Ярославль
2012
ББК 24.23
О 64
УДК 547
Рецензенты: кафедра “Бионеорганическая и биофизическая химия” Ярославской государственной медицинской академии; В.Н.Казин, канд. хим. наук, доцент кафедры общей и биоорганической химии Ярославского государственного университета.
А.Ф.Бетнев, И.С. Колпащикова, Е.Р. Кофанов, Е.М.Алов
О 64 Органическая химия. Часть 1. Углеводороды. Галогенпроизводные углеводородов: Учебное пособие / Яросл. гос. техн. ун-т.- Ярославль, 2012.- 182 с.
ISBN 5-230-17813-2
В пособии рассмотрены основные вопросы строения и реакционной способности, типичные реакции и основные способы получения ациклических, карбоциклических и ароматических соединений, а также галогенпроизводных углеводородов. Пособие составлено с учетом важнейших достижений органической химии за последние годы.
Предназначено для студентов специальности «Химическая технология органических веществ», а также может быть рекомендовано для студентов заочной и дневной формы обучения всех специальностей химико-технологического факультета.
Ил. 18. Табл. 17. Библиогр. 5.
О Без объявл. ББК 24.23
ISBN 5-230-17813-2 ã Ярославский государственный
технический университет, 2012
Введение
Значение химии вообще, и органической химии в особенности, трудно переоценить. Еще в 18 веке великий русский ученый М. В. Ломоносов говорил: «Широко простирает химия руки свои в дела человеческие» и с этим нельзя не согласиться – нет такой отрасли человеческой деятельности, которой бы не коснулись достижения химии.
Органическая химия – это химия соединений углерода. Вначале органическая химия определялась как химия соединений, которые образуются живой материей. Вплоть до середины 19 века многие химики считали, что органические соединения могут образовываться только в живых организмах и, следовательно, их нельзя синтезировать из неорганических веществ.
В настоящее время большинство органических соединений получают синтетически, используя в качестве сырья, в основном более простые органические соединения. Природными источниками простейших органических соединений являются нефть и уголь.
Что же характерно для соединений углерода и заставляет рассматривать их отдельно от соединений остальных ста с лишним элементов периодической системы? Прежде всего, число органических соединений во много раз превышает число неорганических соединений. А молекулы органических соединений могут быть очень большими по размеру и сложными по строению.
Каковы же особенности углерода, позволяющие ему образовывать столь большое число соединений? Атомы углерода могут соединяться друг с другом, как не могут соединяться атомы никакого другого элемента. Атомы углерода могут образовывать цепи из тысяч атомов или кольца любого размера; цепи и кольца могут иметь разветвления и перекрестные связи. Углеродные атомы, участвующие в образовании этих цепей и колец, могут быть связаны с другими атомами, в основном с водородом, а также с кислородом, азотом, серой, фосфором, галогенами и другими элементами.
Дата: 2018-12-21, просмотров: 906.