МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Саратовский государственный
медицинский университет
Кафедра факультетской педиатрии
Медицинская генетика
Под редакцией
канд. мед. наук доц. В.И. Горемыкина
Издательство
Саратовского государственного
медицинского университета.
1997
УДК 57(07)
Учебно-методическое пособие содержит перечень тестов, ответы на которые являются необходимым минимумом знаний студентов педиатрических факультетов вузов. Работа с тестами предопределяет самостоятельную активность к познанию, интерес к изучению соответствующих источников информации, приобретению практических навыков у постели больного.
Теоретическая часть методического пособия является “банком данных”, который соответствует тестовому разделу и определяет минимум теоретических знаний и практических навыков раздела медицинской генетики в педиатрии.
Подготовка студентов по разделу медицинской генетики направлена на формирование самостоятельного врачебного мышления и поведения, умений, обеспечивающих обеспечивающих решение профессиональных задач и применения абсолютно необходимых алгоритмов врачебной деятельности по диагностике, лечению, профилактике наследственных болезеней.
Учебно-методическое пособие предназначено для студентов педиатрических факультетов медицинских вузов.
Составители: канд. мед. наук. И.В.Королева; канд. мед. наук. А.А.Протопопов.
Рецензенты: зав. кафедрой госпитальной педиатрии Саратовского государственного медицинского университета проф. А.С.Эйберман;
доцент Новосибирского государственного медицинского института, член комиссии МЗ РФ по преподаванию медицинской генетики, канд. мед. наук О.В.Лисиченко.
Утверждено в качестве учебно-методического пособия Центральным координационно-методическим Советом СГМУ.
Утверждено Всероссийским учебно-методичечским центром по непрерывному медицинскому и фармацевтическому образованию.
4123000000-55 © Саратовский государственный
М медицинский университет,
И 49(03)-97 1997.
ISB N 5-7213-0051-5
Теоретический минимум по медицинской генетике.
“2 х 2 =?”
1.1. Занятие 1. Предмет и задачи медицинской генетики.
Медицинская генетика изучает роль наследственности в возникновении заболеваний. Значение этого раздела общей генетики определяется уже тем, что около 6% населения (по данным ВОЗ) имеет наследственную отягощенность, 5% детей рождается с наследственными и врожденными заболеваниями, при этом 0,7% приходится на долю хромосомных болезней, 1%- моногенных, 1,3% -мультифакториальных, 2% - врожденных пороков ненаследственной природы.
1.1.1. Задачи медицинской генетики:
1 Изучение роли наследственности и среды в возникновении заболеваний;
2 Разработка методов диагностики и лечения наследственных заболеваний;
3 Прогноз в семьях, где имеются наследственные заболевания;
4 Проведение медико-генетического консультирования.
Понятие наследственных болезней
Наследственные болезни - болезни причиной которых являются вредные мутации (стойкие количественные и качественные нарушения) в наследственном аппарате клеток (в гаметах, зиготе, и первых бластомерах).
Понятие о врожденных пороках развития.
Врожденные пороки развития (ВПР) - стойкие морфологические изменения органа или всего организма, выходящие за пределы вариаций их строения.
Изучением врожденных пороков развития занимается тератология (t eratos - урод) - наука об этиологии, патогенезе и диагностике ВПР.
Тератогенный календарь
| Пороки развития | Неделя беременности |
| Мозг | 2-11 |
| Глаза | 3-7 |
| Сердце | 3-7 |
| Конечности | 2-8 |
| Зубы | 6-10 |
| Уши | 6-11 |
| Губы | 5-6 |
| Зев | 10-11 |
| Органы пищеварения | 11-12 |
| Мочевыделительные органы | 6-11 |
| Половые органы | 12-14 |
Этапы клинико-генеалогического метода.
Этап. Анализ родословной.
Анализ родословной - самый ответственный этап клинико-генеалогических исследований. При этом необходимо установить следующее:
1. Является ли данный признак или заболевание единичным или в семье имеется несколько случаев (семейный характер).
2. Выявить лиц, “подозрительных” в отношении данного заболевания, и составить план их обследования для уточнения диагноза.
3. Определить тип наследования и выяснить по какой линии - материнской или отцовской идет передача заболевания.
4. Выявить лиц, нуждающихся в медико-генетическом консультировании, определить степень генетического риска и клинический прогноз для пробанда и его родственников с учетом особенностей заболевания и его генетической характеристики.
5. Разработать план лечения и профилактики, принимая во внимание индивидуальные и семейные особенности заболевания.
Генетический анализ родословных при моногенных заболеваниях:
1. После составления родословной необходимо определить тип наследования признака:
· по локализации гена - аутосомный; сцепленный с полом.
· по отношению доминантности: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный доминантный, Х-сцепленный рецессивный, У-сцепленный.
2 При анализе родословных учитывают характерные черты, присущие каждому типу наследования:
· аутосомно-доминантный:
а) в родословной прослеживается по вертикали; б) у пораженного, как правило болен один из родителей; в) с одинаковой частотой болеют мужчины и женщины; г) при типичном браке (Аа х аа) риск иметь больного ребенка = 50%.
· аутосомно-рецессивный:
а) с одинаковой частотой болеют мужчины и женщины; б) родители пробанда здоровы; в) в родословной прослеживается по горизонтали; г) при типичном браке (Аа х Аа) риск рождения больного ребенка = 25%; д) при наличии кровно-родственного брака риск иметь больного ребенка возрастает.
· Х-сцепленный доминантный:
а) с одинаковой частотой болеют мужчины и женщины; б) у пробанда болен один из родителей; в) у больного отца все сыновья здоровы, а все дочери больны; г) у больной матери равновероятно рождение больной дочери и сына.
· Х -сцепленный рецессивный:
а) чаще болеют мужчины, женщины являются носителем патологического гена; б) сын никогда не наследует заболевание от отца; в) если пораженный пробанд женщина, её отец обязательно поражен; г) при типичном браке (Х*Х х ХУ) 50% дочерей являются носителями патологического гена, 50% сыновей больны.
· У - сцепленный:
а) заболевание отмечается исключительно у мужчин; б) признак передается от отца к сыну.
Близнецовый метод.
Цели близнецового метода :
· используется в решении вопроса о соотносительной роли генотипа и среды в формировании болезни;
· метод контроля по партнеру используется в анализе фенотипической вариации индивидуального генотипа, генетике развития и т.п. (например, в фармакологии с его помощью оценивают эффективность лекарственных препаратов, в психологии - генетику поведения и т.п.)
Сущность близнецового метода заключается в сравнении внутрипарного сходства (конкордантности) в группах монозиготных (MZ) и дизиготных (DZ) близнецов, что позволяет с помощью формулы Хольцингера оценить относительную роль наследственности и среды в формировании признака (болезни).
Гемеллология - наука о близнецах.
Формула Хольцингера 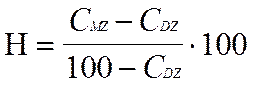
В данной формуле : Н - коэффициент наследуемости;
CMZ - процент конкордантных пар в группе монозиготных близнецов;
CDZ - процент конкордантных пар в группе дизиготных близнецов.
Е - влияние среды вычисляется по формуле Е = 100-Н;
Коэффициент наследуемости (Н) - показатель соотносительной роли наследственности и среды в формировании признака (болезни).
Для всех моногенных заболеваний Н = 100%;
Хромосомных - Н = 100%.
Для мультифакториальных заболеваний - 50% < H < 100%
(сахарный диабет Н=57%, ревматизма -36%, шизофрении - 65%, IQ =80%).
Популяционный метод.
Цели популяционного метода:
· изучение генетической структуры популяции;
· изучение закономерностей распространения патологического признака в популяции;
· анализ встречаемости патологических генотипов и генов в популяциях различных местностей.
Генетическая структура популяции характеризуется частотой генотипов, контролирующих альтернативные вариации признака.
Генофонд характеризуется частотой аллелей данного локуса.
Частота определенного генотипа в популяции определяется количеством особей, обладающих данным генотипом.
Структура популяции зависит:
1. От частоты аллелей в популяции предшествующего поколения;
2. От мутационного давления ( в результате мутационного давления происходит приток аллелей в популяцию);
3. От естественного отбора ( мутантный ген приводит к снижению жизнеспособности, имеющие такой ген умирают не оставляя потомства, в результате происходит отток аллелей из популяции);
4. От типа брака (панмиксия - случайное вступление в брак без учета генотипа, неслучайный подбор положительный - со схожим генотипом: глухие, слепые; неслучайный подбор отрицательный - с противоположным генотипом).
Основная закономерность, позволяющая исследовать генетическую структуру популяций, была установлена в 1908 году независимо друг от друга английским математиком Г. Харди и немецким врачом В. Вайнбергом.
Закон Харди - Вайнберга гласит, что в популяции при условии панмиксии и при отсутствии мутационного давления и отбора устанавливается равновесие частот генотипов, которое сохраняется из поколения в поколение.
С точки зрения популяционного генетического анализа особенно важно то, что закон Харди - Вайнберга устанавливает математическую зависимость между частотами генов и генотипов. Эта зависимость основывается на простом математическом расчете. Если генофонд популяции обусловлен парой аллельных генов, например А и A1 и ген А встречается с частотой р, а ген A1 - с частотой q, то соотношение частот этих аллелей в популяции окажется равенством :
pA + qА1 = 1.
Возведя обе части равенства в квадрат, имеем (pA + qÀ1)2=12, а после раскрытия скобок получим формулу, отражающую частоту генотипов:
p2AA + 2pqAÀ1 + q2À1А1 = 1.
Следовательно, генотип АА встречается в рассматриваемой популяции с частотой p2, генотип A1A1 - с частотой q2, а гетерозиготы - с частотой 2pq. Таким образом, зная частоту аллелей, можно установить частоту всех генотипов, и, наоборот, зная частоту генотипов - установить частоту аллелей.
1.3. Занятие 3. Семиотика наследственной патологии. Анализ фенотипа больного.
Ежегодно описывается несколько сотен новых наследственно обусловленных аномалий. Так по данным каталога W. MCKusik в 1988 году было известно около 3000 наследственных синдромов, а по данным последнего каталога зарегистрировано 4354 наследственных синдрома.
На сегодняшний день многие из наследственных заболеваний диагностируются лишь при анализе совокупности клинических симптомов. В связи с этим в практической деятельности врача педиатра большое значение имеет знание семиотики наследственных болезней.
Положения феногенетики
Феногенетика - раздел медицинской генетики, изучающий семиотику наследственных болезней. Основоположниками феногенетики являются Хейгер и Болеви, сформулировавшие основные признаки, присущие всем наследственным болезням.
У членов одной семьи.
При анализе этого признака ведущая роль отводится клинико-генеалогическому методу, поскольку хорошо собранный семейный анамнез, грамотный анализ родословный помогает врачу в диагностике наследственного заболевания.
При этом необходимо учитывать как агрегацию патологического признака в семье, так и сегрегацию (расщепление) этого признака у членов родословной. Так для синдрома Марфана в классическом варианте характерна следующая триада: поражение опорно-двигательного аппарата, сердечно-сосудистой системы, подвывих хрусталика. Однако у части родственников выявляется только умеренный сколиоз, у других лишь пролапс митрального клапана, тогда как у больного пробанда может наблюдаться полный симптомокомплекс, причем ярко выраженный.
В связи с этим, при анализе родословный врач обязан учитывать пенетрантность и экспрессивность мутантного гена.
Пенетрантность - частота проявляемости гена в признаке, т.е. число (%) особей в популяции, несущих патологический ген и имеющих его клиническое проявление.
Экспрессивность - степень фенотипического проявления гена в признаке. Экспрессивность определяет тяжесть заболевания.
Карта фенотипа.
Фамилия___________ Имя ______________Возраст больного ____________
Масса _____________ рост_______________ телосложение ______________
КОЖА: без изменений, характер изменений __________________________
__________________________________________________________________
рубцы ____________________________________________________________
птеригиум ________________________________________________________
НОГТИ: без изменений, характер изменений _________________________
___________________________________________________________________
ПОДКОЖНАЯ КЛЕТЧАТКА: без изменений, характер изменений ___________________________________________________________________
МЫШЦЫ: без изменений, характер изменений _______________________
___________________________________________________________________
мышечный тонус ___________________________________________________
ЧЕРЕП: окружность головы _________, форма _________; лоб _________;
затылок ____________________;рост волос на лбу _____________________;
головной показатель (ГП) = поперечный размер (расстояние между теменными точками) продольный размер (расстояние м/у точками глабеллы и затылочной костью х 100. У новорожденных ДОЛИХОЦЕФАЛИЯ если ГП < 78 у взрослых - 76. БРАХИЦЕФАЛИЯ - у новорожденных ГП= 83 и >,у взрослых ГП = 81 и >.
УШНЫЕ РАКОВИНЫ: продольный размер ____________; поперечный размер ___________; расположение _________________; деформация ________________________; другие изменения_________________________ __________________________________________________________________.
ОБЛАСТЬ ГЛАЗ И ГЛАЗНОЕ ЯБЛОКО: расстояние между внутренними углами глаз ________; МОИ (межорбитальный индекс = расстояние между внутренними углами глаз/окружность головы на уровне глаз х 100) ______________________________________________________________.
ГИПЕРТЕЛОРИЗМ= МОИ=6,8 и >.ГИПОТЕЛОРИЗМ- МОИ= 4 и <_________________________________________________________________.
Брови и надбровные дуги ____________________. Веки ________________.
Ресницы ___________. Глазная щель __________. Глазное яблоко ______.
НОС: длина носа ______________; ширина __________; без изменений; характер изменений _______________________________________________
ГУБЫ И ПОЛОСТЬ РТА: ширина рта _____________; длина фильтра __________________________________ без изменений; характер изменений ___________________________________________________________________
ВЕРХНЯЯ И НИЖНЯЯ ЧЕЛЮСТИ: без изменений; характер изменений верхней челюсти _______________________________________________
характер изменений нижней челюсти ________________________________
ЗУБЫ: без изменений; прикус _____________________; характер изменений ____________________________________________________________
ЯЗЫК: без изменений; характер изменений ______________ уздечка языка __________________________________________________________________
НЁБО без изменений; характер изменений ________________________
__________________________________________________________________
ШЕЯ: без изменений; характер изменений ___________________________
ГРУДНАЯ КЛЕТКА: окружность ________________; расстояние между сосками __________; без изменений; форма ___________________________; другие изменения __________________________________________________ ___________________________________________________________________
молочные железы __________________________________________________
ПОЗВОНОЧНИК: без изменений; характер изменений ________________ ___________________________________________________________________
ЖИВОТ И ТАЗ: без изменений; характер изменений __________________
___________________________________________________________________
грыжи ____________________________________________________________
ВЕРХНИЕ КОНЕЧНОСТИ: длина ____________; без изменений;
плечо _____________________________________________________________
предплечье ________________________________________________________
ладонь: длина ___________________; изменения _______________________
пальцы: длина среднего пальца ____________; без изменений; характер изменений ________________________________________________________
суставы ___________________________________________________________
НИЖНИЕ КОНЕЧНОСТИ: длина ____________; без изменений;
бедро _______________________; голень ______________________________
стопа _______________________; пальцы _____________________________
___________________________; суставы ______________________________
СЕРДЕЧНО - СОСУДИСТАЯ СИСТЕМА: без изменений; характер изменений __________________________________________________________
ОРГАНЫ ДЫХАНИЯ: без изменений; характер изменений
__________________________________________________________________
ЖЕЛУДОЧНО - КИШЕЧНЫЙ тракт: без изменений; характер изменений ______________________________________________________________
ПЕЧЕНЬ: ________________________________________________________
СЕЛЕЗЕНКА: _____________________________________________________
МОЧЕВАЯ СИСТЕМА: без изменений; характер изменений
___________________________________________________________________
ПОЛОВЫЕ ОРГАНЫ: без изменений; характер изменений
___________________________________________________________________
ЖЕЛЕЗЫ ВНУТРЕННЕЙ СЕКРЕЦИИ: без изменений; характер изменений _____________________________________________________________
ДЕРМАТОГЛИФИКА: без изменений; ладонные складки _____________
осевой трирадиус __________________________________________________
узоры пальцев _____________________________________________________
Фенилкетонурия.
(классификация дана по Mc Kusik V.A., 1988)
ФЕНИЛКЕТОНУРИЯ 1.
Классическая фенилкетонурия (ФКУ) описана А. Folling.,1934 г.
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12 хромосомы.
В основе болезни лежит дефицит фермента фенилаланин -4- гидроксилазы, обеспечивающего превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин, и др.
В патогенезе ФКУ имеют значение следующие механизмы:
1. Прямое токсическое действие на ЦНС фенилаланина и его производных;
2. Нарушение в обмене белков, липо- и гликопротеидов;
3. Расстройства транспорта аминокислот;
4. Нарушение метаболизма гормонов;
5. Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
6. Нарушение функции печени : диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота классической ФКУ среди новорожденных по данным массового скрининга в среднем колеблется от 1 : 5000 до 1 : 10000 по разным регионам России.
ФЕНИЛКЕТОНУРИЯ 2.
Впервые атипичная ФКУ описана I. Smith, 1974 г. Заболевание связано с дефицитом дигидроптеридинредуктазы.
Заболевание наследуется аутосомно-рецессивно. Генный дефект локализуется в коротком плече 4 хромосомы, участке 4р 15.3.
В результате недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина, и триптофана.
Частота заболевания составляет 1 : 100 000 новорожденных.
Рано начатое лечение способствует нормализации фенилаланина в крови, однако не предупреждает появление клинической симптоматики, которая развивается в начале второго полугодия жизни. Фенилкетонурию 2 называют диеторезистентной ФКУ.
ФЕНИЛКЕТОНУРИЯ 3.
Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин синтетазы, участвующей в процессе синтеза тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ 2.
Частота болезни составляет 1 : 30 000 новорожденных.
Фенилкетонурия 3 также диеторезистентна
ДРУГИЕ ВАРИАНТЫ ФКУ.
Эти формы ФКУ связаны с нарушением альтернативных путей обмена фенилаланина. Формируется метилминдальная ацидурия и парагидроскифенилуксусная ацидурия.
МАТЕРИНСКАЯ ФЕНИЛКЕТОНУРИЯ.
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Патогенез мало изучен, предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Так как эмбрион особенно чувствителен к тератогенным воздействиям , рекомендуется начинать диету еще до наступления беременности. В суточном рационе использовать менее 15-20 мг/кг фенилаланина. При это важно избегать дефицита незаменимых аминокислот.
КЛИНИКА ФЕНИЛКЕТОНУРИИ.
При рождении больные фенилкетонурией не отличаются от других новорожденных. Манифестация ФКУ происходит обычно в возрасте 2-6 месяцев.
Первыми проявлениями болезни служат:
· вялость ребенка, отсутствие интереса к окружающему;
· повышенная раздражительность, беспокойство;
· срыгивание, рвота;
· судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
· судорожный синдром;
· заплесневелый, мышиный, волчий запах мочи и пота.
При отсутствии лечения формируется задержка статико-моторного и психоречевого развития, умственная отсталость достигает, как правило, глубокой степени.
Характерны такие фенотипические особенности, как экзематозные и себорейные сыпи, гипопигментация кожи, волос, радужной оболочки глаз.
ДИАГНОСТИКА ФЕНИЛКЕТОНУРИИ.
ФКУ может быть диагностирована на основе обнаружения следующих признаков:
· стойкой гиперфенилаланинемии (более 240 ммоль/л);
· вторичного дефицита тирозина;
· экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
В настоящее время, согласно приказу Минздрава России № 316 от 30.12.93 проведение неонатального скрининга на ФКУ стало обязательным. Скрининирующие тесты должны быть простыми, недорогими и информативными. Этим требованиям отвечают методы, используемые для ранней диагностики ФКУ:
· микробиологический тест Гатри;
· метод флюоресцирующих антител (лабораторный комплекс “Флюороскан”, позволяющий проводить 800 проб в час);
· метод тонкослойной хроматографии .
Оптимальные сроки обследования новорожденных - доношенных, зрелых - 5-6 день жизни; недоношенных, незрелых, больных - 10-14 день жизни.
Трактовка результатов:
1 группа - уровень фенилаланина не превышает 200 ммоль/л (1-3 мг%) - норма;
2 группа - уровень фенилаланина составляет 200-500 ммоль/л (3-10 мг%) - гиперфенилаланинемия. В эту группу входят дети с транзиторной гиперфенилаланинемией, вследствие незрелости ферментных систем печени и больные ФКУ. За данной группой проводится наблюдение по следующему плану: если в течение 6 недель при еженедельном исследовании уровень фенилаланина остается менее 500 ммоль/л, контроль за уровнем фенилаланина крови проводят до 1 года первоначально каждые 3 месяца, а затем каждые 6 мес. жизни. При уровне более 500 ммоль/л назначается диетотерапия.
3 группа - уровень фенилаланина превышает 500 ммоль/л (более 10 мг%), диагностируется ФКУ и с момента постановки диагноза назначается диетотерапия.
В настоящее время разрабатываются и внедряются молекулярно-генетические методы диагностики генного дефекта при ФКУ. Прямая диагностика мутантного гена проводится с помощью синтетических олигонуклеотидных зондов, этот метод пригоден для дородовой диагностики ФКУ и выявления гетерозиготного носительства.
Помимо молекулярно-генетического анализа, выявление гетерозигот может осуществляться биохимическими тестами после нагрузки фенилаланином в дозе 25 мг/кг.
Диеторезистентные формы ФКУ диагностируют при помощи:
· исследования биоптеринов мочи ;
· перорального нагрузочного теста с тетрагидробоиптерином (через 4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит резкое снижение и нормализация уровня фенилаланина в крови с одновременным повышением уровня тирозина);
· исследования активности дигидроптеридинредуктазы и 6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов, эритроцитах, гепатоцитах.
ЛЕЧЕНИЕ ФЕНИЛКЕТОНУРИИ.
1 Главным способом лечения является диетотерапия, ограничивающая поступление в организм фенилаланина; приступить к ней нужно немедленно после установления диагноза.
Из рациона больных исключаются: молоко, молочные продукты, творог, мясо, мясные продукты, колбасы, рыба, яйца, хлебобулочные изделия, фасоль, горох, орехи, шоколад.
Белковым эквивалентом этих продуктов питания становятся гидролизаты белка, либо аминокислотные смеси, лишенные фенилаланина: “Лофенолак”, “Фенилфри” (США), “Берлофен”, “Апонти”, “Гипофенат” у детей до 4-5 лет и “Нофелан” - у детей старше 5 лет.
В пищевой рацион входят овощи, фрукты, мед, растительное масло, безбелковый хлеб.
Наиболее рационально отменять диетическое лечение в возрасте 7-8 лет.
2 Препараты с промедиаторным действием:
· Наком (комбинация карби ДОФА и лево ДОФА) - доза 100-375 мг/сутки в течение 3-4 недель, перерыв между курсами 1,5-2 месяца.
· Лево-дофа - доза 10-15 мг/кг в сутки;
· 5- окситриптофан - доза 10 мг/кг сутки.
Ноотропные препараты.
Витамины.
ЛФК, массаж.
6 Диеторезистентные формы. Лечение включает назначение тетрагидробиоптерина - доза 10-20 мг/кг в сутки.
Синдром Марфана
Синдром Марфана (СМ) - системное наследственное заболевание соединительной ткани. Впервые описано Вильямсом в 1876 г.
Тип наследования - аутосомно-доминантный, с высокой пенетрантностью гена. Частота = 1 : 25 000.
Локализация гена - в длинном плече 15 хромосомы, поле 21. Суть мутации - замена в белке фибриллина пролина на аргинин. В результате происходит повышение синтеза коллагена типа 3 и уменьшение содержания коллагена типа 1, в норме соотношение коллаген 1 : коллаген 3 = 6 : 4; а при СМ = 3 : 7.
Клиническая картина.
1 Скелетные аномалии : длинные тонкие конечности, арахнодактилия кистей и стоп, воронкообразные или килевидные деформации грудной клетки, кифосколиозы, узкий лицевой скелет, “готическое” небо, астеническое телосложение, высокий рост (выше 97 перцентиля). Оценка арахнодактилии :
· рентгенологическая по метакапальному индексу (отношение длины к ширине 2-5 метакарпальных костей), вычисляемого по правой кисти. При СМ метакарпальный индекс = 8,0-11,0 (норма =6,4-7,9);
· “положительный симптом большого пальца”;
· “положительный симптом запястья”.
2 Миотонический синдром: гипермобильность (“разболтанность”) суставов, плоскостопие, вывихи, подвывихи.
3 Поражение сердечно-сосудистой системы: пролапсы клапанов, чаще митрального; аневризмы аорты, нарушения в проводящей системе сердца (нарушение проводимости по правой ножке пучка Гиса), вторичные метаболические кардиомиопатии.
4 Поражение органов зрения: вывихи и подвывихи хрусталика, вследствие слабости цинновой связки, миопия или гиперметропия, страбизм, астигматизм, сферофокия, иридодонез, может быть отслойка сетчатки, вторичная глаукома, расширение вен глазного дна, дегенерация сетчатки.
5 Поражение центральной нервной системы: могут формироваться артерио-венозные аневризмы в головном мозге, кисты. У некоторых больных отмечается снижение интеллекта.
6 Поражение бронхолегочной системы: бронхоэктазы, затяжные обструктивные бронхиты, ателектазы, трахеобронхомегалия, эмфизема, поликистоз.
7 Поражение мочевыделительной системы: нефроптоз, вторичный пиелонефрит, бессимптомная гематурия.
Вегето-сосудистые дистонии.
Остеохондрозы.
ЛЕЧЕНИЕ СИНДРОМА МАРФАНА
· Диетотерапия. Применение продуктов с высоким содержанием НЭЖК. Включение высокожировых энпитов, пищи, обогащенной витаминами А, Е, В, С.
· По показаниям - для снижения МО бета-адреноблокаторы (обзидан и пр.)
· Препараты калия: панангин, аспаркам.
· Ноотропы.
· Витамины (А, В, С, Е и пр.)
Диспансеризация больных СМ.
· Врач педиатр, терапевт, кардиолог, окулист, хирург-ортопед, стоматолог, ЛОР-врач, психотерапевт осматривают 2 раза в год.
· ЭКГ, ФКГ, ЭХО-кардиография - 1 раз в год.
· Препараты нормализующие обмен (витамины и пр.), ноотропы - 2 раза в год курсами.
· По показаниям курсы обзидана 2 раза в год.
· Противопоказаны занятия требующие больших физических и эмоциональных нагрузок (рекомендовать ЛФК, занятия в спец. группах).
Муковисцидоз
Муковисцидоз - генерализованная экзокринопатия с преимущественным поражением легких и желудочно-кишечного тракта.
Заболевание характеризуется системным поражением экзокринных желез, причем как слизеобразующих (респираторного тракта, кишечника, поджелудочной железы), так и серозных (слюнных, потовых, слезных).
Первое описание болезни дал Петер Пауф в 1595 году.
Частота муковисцидоза : 1 : 2000 (ВОЗ), 1 : 26000 (Финляндия), 1 : 1250 (Франция).
Тип наследования аутосомно-рецессивный.
Локализация гена в длинном плече 7 хромосомы, сегменте 31. Ген включает 250 тыс. нуклеотидных пар, кодирует синтез белка из 1480 аминокислот, Продукт гена - мембранный транспортный белок (МТРБ), функция которого - перенос ионов через мембраны эпителиальных клеток. Открыто 230 мутаций гена муковисцидоза - возможны 230- вариантов клиники. В 70% случаев муковисцидоза имеет место точковая мутация гена - делеция нуклеотидов, определяющих положение фенилаланина - 508 аминокислоты в полипептидной цепи. Мутация маркирована как DF 508. Возможно её пренатальное определение.
Патогенез. При муковисцидозе имеется сочетание дефекта реабсорбции натрия хлорида на уровне экскреторных канальцев слизистых и потовых желез с увеличением содержания сиаломуцинов в слизистом секрете, ведущим к увеличению вязкости последнего, закупорке выводных протоков желез, затруднению эвакуации слизи.
БОЛЬНЫХ НА МУКОВИСЦИДОЗ
· Мекониальный илеус у новорожденных;
· Частые заболевания бронхолегочной системы:
а) рецидивирующие обструктивные бронхиты;
б) затяжные и хронические пневмонии;
в) дыхательные аллергозы;
г) воспалительные заболевания бронхолегочной системы, сопровождающиеся высевом из мокроты Psevdomonas aeruginosa.
· Хронические заболевания желудочно-кишечного тракта:
а) диспептические нарушения, плохая переносимость жиров, большое количество нейтрального жира в кале;
б) колиты;
в) гастриты и язвенная болезнь;
г) цирроз печени неясной этиологии
д) выпадение прямой кишки при дефекации;
е) склонность к запорам;
ж) упорные дисбактериозы.
· Задержка физического развития, плохая прибавка в массе, при хорошем аппетите и склонности к полифекалии;
· Случаи мужского бесплодия в семье (при муковисцидозе нарушается подвижность сперматозоидов вследствие густого секрета простаты);
· Рецидивирующая кишечная непроходимость;
· Частые поражения придаточных пазух носа.
ЛЕЧЕНИЕ МУКОВИСЦИДОЗА.
1. Диета высококалорийная, с увеличением потребления белка до 3-5 г/кг массы тела в сутки. Хорошо добавлять в рацион высококалорийные энпиты и детские смеси.
2. Заместительная терапия:
· ферменты (панкреатин - до 16,0 в сутки; креон, нео-панпур, панзитрат, панкурмен, дигестал, мезим форте, панзинорм, фестал);
· Фросколин 300 - увеличивает количество хлорных канальцев на мембране;
· Амилорид блокирует выведение ионов хлора в секрет;
· АТФ, УТФ - активизируют ТРБМ.
3. Санация трахеобронхиального дерева:
· Муколитики (ацетилцистеин, мукомист, мукосольвин, амброксол, бромгексин, мукодин, мистаброн - в/м, перорально, в ингаляциях)
· Постуральный дренаж, вибрационный массаж;
· Кашель мелкими толчками;
· Санационные бронхоскопии.
4. Подавление инфекционного процесса. Учитывая характер высеваемой флоры (синегнойная палочка) назначают цефалоспорины 3 генерации (клафоран, цефотаксим, цефобид, фортум, лонгацеф; группу карбапенемов (тиенам); аминогликозиды 3 генерации (бруламицин, небцин, амикацин, нипоцин); фторхинолоны (таривид, абактал, ципробай, ципробид, ципрофлоксацин).
5. Витамины: А, Е, С.
6. Физиолечение в период обострения воспалительного процесса в легких применяют УВЧ, УКВ, электрофорез препаратами магния, йода; широко пользуются ингаляциями.
7. Хирургическое лечение должно проводиться лишь по витальным показаниям ( пневмоторакс, пиопневмоторакс, кишечная непроходимость); хирургическое лечение бронхоэктазов при муковисцидозе не оправдано.
ПРОФИЛАКТИКА МУКОВИСЦИДОЗА
Пренатальная диагностика:
а) биохимическими методами -определение на 17-18 неделе беременности в амниотической жидкости снижения содержания ферментов микроворсинок кишечника (гамма - глутамилтранспептидазы, аминопептидазы, щелочной фосфатазы).
б) молекулярно - генетическими методами: прямая детекция мутантного гена, выявление мутантного гена при помощи ДНК маркеров.
2. Медико-генетическое консультирование семей высокого риска.
3 Активное выявление больных муковисцидозом: внедрение программ массового скрининга новорожденных (определение в крови содержания иммунореактивного трипсина, определение мутации DF 508);
Синдром Нунан
Минимальные диагностические признаки: крыловидные складки на шее, аномалии грудной клетки, крипторхизм, врожденные пороки сердца (фенотип, напоминающий синдром Тернера при нормальном кариотипе).
Клиническая характеристика: Характерными чертами лица являются гипертелоризм, эпикант, антимонголоидный разрез глаз, птоз, микрогнатия, низко посаженные уши, арковидное небо. Отмечаются миопия, косоглазие, кератоконус. Шея толстая, короткая, с крыловидными складками. Приблизительно 80% сердечных изменений составляют пороки правого сердца. В 27% случаев наблюдаются пороки мочевыводящей системы (гидронефроз, удвоение лоханок, гипоплазия почек). Часто имеет место гирсутизм, поперечная ладонная складка. Функция половых желез варьирует от полного агонадизма до сохранения фертильности.
Умственная отсталость наблюдается в 61% случаев.
Тип наследования: предположительно аутосомно - доминантный .
Соотношение полов - М1 : Ж1.
Нейрофиброматоз
Синдром Меккеля
Минимальные диагностические признаки: черепно-мозговая грыжа, полидактилия, поликистоз почек.
Клиническая характеристика: Наиболее важным диагностическим признаком синдрома Меккеля является затылочная черепно-мозговая грыжа (80%). Кроме того, отмечается микроцефалия, гидроцефалия, гипоплазия мозолистого тела, аринэнцефалия. Описывают скошенный лоб, низкорасположенные деформированные ушные раковины, микрогению, гипертелоризм, расщелину неба. У 25% детей имеются пороки развития глаз: микрофтальмия, колобомы, катаракта.
Поликистоз почек отмечается в 75%, могут быть другие пороки мочевой системы - атрезия мочеточников, гипо- или аплазия мочевого пузыря. Гонады уменьшены и диспластичны, для мальчиков характерны крипторхизм, гипоплазия наружных половых органов, для девочек- двурогая матка, атрезия влагалища, перегородка влагалища.
Характерным признаком является полидактилия, чаще шестипалость, которая может сочетаться с синдактилией.
Популяционная частота - 1 : 65 000.
Соотношение полов - М1 : Ж1.
Тип наследования - аутосомно-рецессивный.
Синдром Холта - Орама
Синдром Корнелии Де Ланге
Минимальные диагностические признаки: задержка психо -моторного развития, микроцефалия, синофриз, длинный фильтр, вывернутые наружу ноздри, тонкая загнутая внутрь верхняя губа, микромелия, гирсутизм.
Клиническая характеристика: Аномалии головы и лица придают больному своеобразный вид - микробрахицефалия, деформированные ушные раковины, синофриз, длинные загнутые ресницы, миопия, атрофия или колобома зрительного нерва, маленький нос с вывернутыми ноздрями, высокое арковидное небо. Характерными пороками конечностей являются микромелия, ограничение подвижности локтевых суставов, фокомелия и олигодактилия.
Наблюдаются гипертрихоз, мраморная кожа, гипоплазия сосков.
В 100% случаев отмечается умственная отсталость задержка роста, мышечный гипертонус, слабый высокий голос. Описаны пороки внутренних органов: врожденные пороки сердца, поликистоз, гидронефроз, удвоение почек, неполный поворот кишечника, пилоростеноз, диафрагмальная грыжа.
Популяционная частота - 1 : 12 000.
Соотношение полов - М1 : Ж1.
Тип наследования - неизвестен.
1.5. Занятие 5. Хромосомные болезни.
Хромосомные болезни - наследственные заболевания, генетической основой которых являются отклонения от нормального содержания в клетках организма количества хромосомного материала.
Частота - Ежегодно в России рождается около 30 тысяч детей с хромосомной патологией. Среди всех новорожденных аномалии в хромосомном наборе имеют 0,7%, среди недоношенных детей - 2,5%. Мертворождения являются результатом хромосомной патологии в 7,2% случаев, спонтанные выкидыши - в более чем 50%.
Номенклатура
Стандартизирована номенклатура для написания кариотипа. Во-первых, записывается общее число хромосом, после чего следует описание аберраций. Любое увеличение хромосомного материала обозначают знаком “+”, потерю - знаком “-“. Знак помещается перед номером хромосомы, если речь идет о целой хромосоме, и после символа, обозначающего плечо. Хромосомы с транслокацией записывают в скобках, перед которыми ставят литеру “t”.
Примеры записи кариотипа:
·47, ХХ + 13 - женщина с трисомией 13 (синдром Патау);
·47, ХУ + 18 - мужчина с трисомией 18 (синдром Эдвардса);
·47, ХУ + 21 - мужчина с трисомией 21 (синдром Дауна);
·46, ХУ - 21, +t (21q 21q) - мужчина с синдромом Дауна в результате транслокации типа центрического соединения между двумя хромосомами 21, замещение одной хромосомой 21;
·45, ХХ -14, -21, +t (14q 21q) - фенотипически нормальная женщина - носитель транслокации типа центрического соединения между хромосомами 14 и 21;
·46, ХУ, del (5p) - мужчина с синдромом “кошачьего крика”, обусловленным делецией части короткого плеча хромосомы 5;
·45, ХО - женщина с синдромом Тернера-Шерешевского, обусловленным моносомией Х;
·46, ХУ / 47, ХХУ - мужчина с мозаичным синдромом Клайнфельтера.
1.5.4. Классификация хромосомных болезней:
1 Аномалии аутосом (геномные и хромосомные);
2 Аномалии половых хромосом (геномные и хромосомные).
Синдром Клайнфельтера
Частота - 1: 1000 новорожденных мальчиков.
Кариотип 47, ХХУ, редкие варианты: 48, ХХХУ; 49, ХХХХУ; возможен мозаицизм 46, ХУ/47, ХХУ.
Фенотипическая характеристика:
· Клиническая манифестация в пре- и пубертатный период. Не происходит увеличение яичек, а у некоторых отмечается тенденция к их уменьшению и уплотнению, формируется микроорхидизм. Гистологически доказанное нарушение сперматогенеза в форме азооспермии. Размеры полового члена обычные или несколько уменьшенные. Бесплодны. Потенция снижена.
· Снижение интеллекта - IQ от 85% до 50%. Особенности психики: снижение эмоционально-волевой сферы, внушаемы, неусидчивы, подвержены влиянию, конфликтны, нет быстрой реакции (учитывать при профориентации). В связи с этим важно активно выявлять таких больных перед поступлением в школу, вести индивидуально совместно врачу, педагогу, психологу. При отсутствии этого отмечается склонность к асоциальному поведению.
· Высокий рост;
· Преобладание нижнего сегмента тела над верхним:, длинные ноги, ширина таза больше ширины плеч;
· Гинекомастия, у части больных истинная за счет развития ткани молочной железы, могут быть опухоли;
· На лице и теле оволосение скудное, на лобке развито по женскому типу.
· Брахицефалия, низкий рост волос на затылке, клинодактилия 5, сколиоз.
Расчет генетического риска: Риск для сибсов в спорадических случаях(при нормальном кариотипе родителей) не превышает популяционного.
Понятие предрасположенности
Реализация внешних факторов в болезнь возможна при наличии предрасположенности к той или иной патологии. Предрасположенность - это изменение нормы реакции организма на действие факторов внешней среды. Предрасположенность определяется индивидуальной вариацией генов, что, в свою очередь, придает индивидуальность строения органов, физиологическим функциям, уровню ферментов и пр. В результате одно и то же по интенсивности внешнее воздействие для одного индивидуума - безвредна; а у другого приводит к формированию болезни.
Имеются ряд особенностей детского организма, делающих его более подверженным МФЗ:
· незрелость центральных регуляторных механизмов;
· повышенная проницаемость кожи и слизистых;
· незрелость общего и местного иммунитета;
· незрелость ферментных систем печени;
· низкая клубочковая фильтрация.
Развитие организма следует рассматривать как реализацию наследственной программы, заложенной в зиготе и передаваемой из поколения в поколение, но на реализацию этой программы воздействуют различные факторы на разных этапах развития организма.
Этиология МФЗ
Таким образом, можно условно выделить три составляющих в этиологии МФЗ : генетическую, онтогенетическую, средовую.
Патогенез МФЗ
В самом общем виде патогенез МФЗ объясняется мутациями множества генов, которые действуют однонаправленно, дополняя друг друга - т.е. аддитивно. Возможно преимущественное влияние какого-либо одного гена. Патологические свойства этих генов проявляются только при воздействии неблагоприятных факторов внешней среды. Относительная роль наследственности и среды различна не только для каждой болезни, но и для каждого индивидуума в отдельности.
Общие черты МФЗ
·наличие патогенетических и ассоциативных маркеров предрасположения;
· хроническое течение с наличием “клинического континуума” (непрерывный ряд форм различной выраженности - от субклинических до манифестных);
· более раннее начало и утяжеление клинической картины в нисходящих поколениях семьи;
· повышение риска рождения больного ребенка в каждом последующем поколении;
· однотипность проявлений болезни у ребенка и ближайших родственников;
· повышенный риск повторного рождения предрасположенных к болезни детей с появлением каждого последующего ребенка.
В противоположность моногенным доминантным и рецессивным признакам - риск для родственников сильно варьирует в разных семьях. Он заметно возрастает в семьях, где уже есть два пораженных члена по сравнению с семьями, где имеется только один пораженный. Более тяжелая степень выраженности дефекта у пробанда сопровождается повышенным риском поражения для родственников.
Для многих МФЗ характерны половые различия в частоте поражения, при этом более высокий риск заболевания имеют близкие родственники тех пробандов, которые принадлежат к редко поражаемому полу.
Маркеры МФЗ
Маркеры (метки) МФЗ - признаки, указывающие на наличие генетической составляющей в возникновении болезни. Выделяют ассоциативные и патогенетические маркеры. К ассоциативным маркерам относятся группы крови (системы АВО), антигены главного комплекса гистосовместимости по системе HLA. Так, например язвенная болезнь чаще встречается у детей с 1 группой, ревматизм - со 2-й. Одно из наиболее обоснованных объяснений для механизма действия всех ассоциативных маркеров - плейотропное действие генов, т.е. одновременное воздействие генов и на маркер и на ряд биохимических, иммунологических и физиологических параметров, изменения которых оказываются характерными (маркерными) именно для этого заболевания. Возможно “сцепление” локусов генетического маркера и заболевания. Один и тот же ассоциативный маркер может быть для различных заболеваний.
С практических позиций более перспективен поиск биологических (патогенетических) маркеров предрасположения, т.е. маркеров, непосредственно отражающих патогенез болезни. Наличие взаимосвязи между клиникой заболевания и патогенетическим маркером означает, что определенная часть генных локусов, вовлеченных в генетическую систему заболевания, является общей и для какого-либо маркерного иммунологического или биохимического признака. Патогенетические маркеры делятся на одномерные - когда один какой-либо показатель указывает на болезнь и многомерными - когда о наличии патологии свидетельствует комплекс показателей. Например для атеросклероза биохимическими маркерами являются гиперхолестеринемия, гипертриглицеридемия.
Аллергические заболевания
Имеются многочисленные факты подтверждающие высокую степень генетического участия в возникновении аллергических заболеваний. Если аллергия имеется у обоих родителей то риск какого-либо аллергического заболевания составляет 40-60%; риск максимален (80%) если у обоих родителей поражен один и тот же орган мишень (например бронхиальная астма или атопический дерматит). В целом у детей первых трех лет жизни с отягощенной по аллергическим заболеваниям наследственностью частота таких болезней в 10 раз выше, чем у детей с благоприятной наследственностью. Симптомы аллергических болезней появляются тем раньше, чем сильнее отягощен анамнез. У членов одной семьи чаще поражается один и тот же орган.
Доказан генетический контроль за синтезом Ig E, играющего решающую роль в возникновении аллергических заболеваний. По крайней мере 2 гена конторолируют синтез Ig E: так называемый ген атопии и Ir - ген, который ответственен за продукцию Ig E и синтез других Ig. Склонность к гиперпродукции Ig E передается как полигенно, так и как аутосомно-рецессивный признак. Генетический контроль осуществляется и за другими звеньями аллергического процесса - за активностью макрофагов и мастоцитов, их способностью высвобождать медиаторы, за проницаемостью слизистых оболочек для различных АГ и др. В пределах мультифакториальной генетической системы существуют как факторы общего характера, т.е. усиливающую общую подверженность к атопическим заболеваниям, так и действующие совместно с ними другие факторы, влияющие на поражение конкретных органов. Для возникновения аллергической патологии одной лишь генетической предрасположенности недостаточно. Необходимо параллельное влияние разрешающих экзогенных факторов: попадание аллергена, мембрандестабилизирующих факторов и пр. Примером МФЗ аллергической природы может служить Бронхиальная астма. При которой генетически обусловлены: повышение общего и специфического IgE; низкий уровень гаптоглобина; имеется связь с группой крови А(П); часто выявляются у больных HLA- антигены A3; B7; DW2; С антигенами BW16 связаны наиболее тяжелые формы, с BW21, A9, B7- более легкие формы; гипореактивность бета-2 адренорецепторов; гиперреактивность холинорецепторов бронхов.
Во многом детерминированы генетически хронические и рецидивирующие пневмонии. При этом генетическая составляющая включает в себя нарушение иммунитета (дефицит синтеза Ig A , нарушение функции отдельных субпопуляций или клонов Т-лимфоцитов), нарушение мукоцилиарного транспорта.
Гипертоническая болезнь
Существует отчетливо выраженная генетическая предрасположенность к гипертонической болезни (ГБ). Популяционная вариабельность величины АД у детей примерно на 50% определяется генетическими и на 50% экзогенными факторами. При наличии артериальной гипертензии у обоих родителей вероятность возникновения ГБ у ребенка составляет 57%, если страдает только мать - 30%, только отец - 13%. Коэффициент наследуемости минимального АД = 85%, среднего - 78%.
Патогенез наследственных изменений при ГБ сложен и многоэтапен. Отмечается наследственная повышенная выработка инсулина в ответ на поступление в организм глюкозы, что стимулирует рост и миграцию сосудистых гладкомышечных клеток и приводит в итоге к увеличению периферического сопротивления сосудов и повышению АД. Кроме того, наследуется повышенная симпатоадреналовая активность, склонность к задержке натрия, повышения внутриклеточного кальция, что приводит к неадекватному спазму артерий в ответ даже на нормальный выброс адреналина; более быстрому развитию структурных изменений в стенке сосудов.
Наследуются и психологические особенности личности, способствующие возникновению ГБ - так называемое поведение типа” А” (сочетание сильного типа высшей нервной деятельности с повышенной агрессивностью и склонностью к межличностным конфликтам).
Этап - Уточнение диагноза.
Выбор метода исследования.
1. Моногенные заболевания:
· клинико-генеалогический метод;
· биохимический метод;
· иммунологический метод;
· молекулярно-генетический метод.
2. Хромосомные заболевания:
· клинико-генеалогический метод;
· цитогенетический метод.
3. Мультифакториальные заболевания:
· клинико-генеалогический метод;
· определение моногенно наследуемых маркеров.
4. Фенокопии:
· клинико-генеалогический метод;
· тщательно изучить характер течение беременности;
· цитогенетический (для исключения хромосомных болезней);
· биохимический (для исключения моногенных болезней),
Этап - Оценка риска.
1. Риск до 5% - низкий риск, чаще наблюдается при мультифакториальных заболеваниях.
2. Риск 6-10% - повышенный риск легкой степени, также чаще наблюдается при мультифакториальных заболеваниях.
3. Риск 11-20% - повышенный риск средней степени, наблюдается при хромосомных заболеваниях и моногенных заболеваниях с низкой пенетрантностью гена.
4. Риск более 20% - высокий риск, наблюдается при моногенных заболеваниях и некоторых транслокационных вариантах хромосомных болезней.
ОГЛАВЛЕНИЕ
Медицинская генетика
Учебно-методическое пособие
Редактор Л.А. Алехнович
Подп. к печ. __________ 1997 г. Тираж ________ .
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Саратовский государственный
медицинский университет
Кафедра факультетской педиатрии
Медицинская генетика
Под редакцией
канд. мед. наук доц. В.И. Горемыкина
Издательство
Саратовского государственного
медицинского университета.
1997
УДК 57(07)
Учебно-методическое пособие содержит перечень тестов, ответы на которые являются необходимым минимумом знаний студентов педиатрических факультетов вузов. Работа с тестами предопределяет самостоятельную активность к познанию, интерес к изучению соответствующих источников информации, приобретению практических навыков у постели больного.
Теоретическая часть методического пособия является “банком данных”, который соответствует тестовому разделу и определяет минимум теоретических знаний и практических навыков раздела медицинской генетики в педиатрии.
Подготовка студентов по разделу медицинской генетики направлена на формирование самостоятельного врачебного мышления и поведения, умений, обеспечивающих обеспечивающих решение профессиональных задач и применения абсолютно необходимых алгоритмов врачебной деятельности по диагностике, лечению, профилактике наследственных болезеней.
Учебно-методическое пособие предназначено для студентов педиатрических факультетов медицинских вузов.
Составители: канд. мед. наук. И.В.Королева; канд. мед. наук. А.А.Протопопов.
Рецензенты: зав. кафедрой госпитальной педиатрии Саратовского государственного медицинского университета проф. А.С.Эйберман;
доцент Новосибирского государственного медицинского института, член комиссии МЗ РФ по преподаванию медицинской генетики, канд. мед. наук О.В.Лисиченко.
Утверждено в качестве учебно-методического пособия Центральным координационно-методическим Советом СГМУ.
Утверждено Всероссийским учебно-методичечским центром по непрерывному медицинскому и фармацевтическому образованию.
4123000000-55 © Саратовский государственный
М медицинский университет,
И 49(03)-97 1997.
ISB N 5-7213-0051-5
Теоретический минимум по медицинской генетике.
“2 х 2 =?”
1.1. Занятие 1. Предмет и задачи медицинской генетики.
Медицинская генетика изучает роль наследственности в возникновении заболеваний. Значение этого раздела общей генетики определяется уже тем, что около 6% населения (по данным ВОЗ) имеет наследственную отягощенность, 5% детей рождается с наследственными и врожденными заболеваниями, при этом 0,7% приходится на долю хромосомных болезней, 1%- моногенных, 1,3% -мультифакториальных, 2% - врожденных пороков ненаследственной природы.
1.1.1. Задачи медицинской генетики:
1 Изучение роли наследственности и среды в возникновении заболеваний;
2 Разработка методов диагностики и лечения наследственных заболеваний;
3 Прогноз в семьях, где имеются наследственные заболевания;
4 Проведение медико-генетического консультирования.
Дата: 2019-05-28, просмотров: 328.