Молекулярно-генетические аспекты апоптоза
⇐ ПредыдущаяСтр 14 из 14
Поможем в ✍️ написании учебной работы
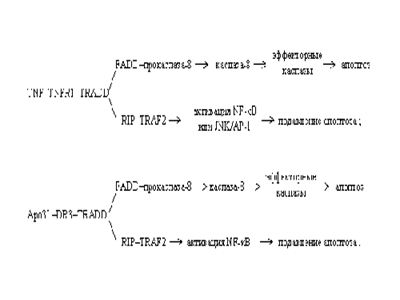 2. Существует путь передачи сигнала апоптоза с участием эндоплазматического ретикулума (ЭР). В ЭР локализована прокаспаза-12. Нарушение внутриклеточного Ca2+-гомеостаза добавкой тапсигаргина или Ca2+-ионофорного антибиотика А23187 ведет к апоптозу клеток, вызванному превращением прокаспазы-12 в каспазу-12. ЭР-зависимый апоптоз связан с болезнью Альцгеймера: кортикальные нейроны мышей, дефицитных по каспазе-12, устойчивы к апоптозу, индуцированному І-амилоидным белком, но не к апоптозу с участием рецепторов плазматической мембраны или митохондриального цитохрома С. 3. Цитотоксические лимфоциты, Т-киллеры, могут вызывать апоптоз у инфицированных клеток с помощью белка перфорина. Полимеризуясь, перфорин образует в цитоплазматической мембране клетки-мишени трансмембранные каналы, по которым внутрь клетки поступают TNFb , гранзимы (фрагментины) - смесь сериновых протеаз. Существенным компонентом этой смеси является гранзим В - протеолитический фермент, превращающий прокаспазу-3 в активную каспазу-3. 4. Высвобождаемый из митохондрий цитохром С вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) участвует в активации каспазы-9. APAF-1 - белок с молекулярной массой 130 кДа, содержащий CARD-домен (caspase activation and recruitment domain) на N-конце и 12 повторяющихся аминокислотных WD-40-последовательностей (WD - дипептид из триптофана и аспартата) на С-конце, образует комплекс с прокаспазой-9 в присутствии цитохрома С и dATP или АТР. К наиболее охарактеризованным WD-белкам относится ?-cубъединица G-белков. Из этих субъединиц собираются жесткие, симметричные структуры, наподобие веера или пропеллера. WD-повторы свойственны белкам, участвующим в регуляции деления и дифференцировки эукариотических клеток, транскрипции генов, модификации мРНК, трансмембранной передачи сигналов, слияния мембранных везикул. Среди прокариот WD-белки обнаружены у цианобактерий. APAF-1 играет роль арматуры, на которой происходит аутокаталитический процессинг каспазы-9. Предполагается, что в результате зависимого от гидролиза dATP (или АТР) конформационного изменения APAF-1 приобретает способность связывать цитохром С. Связав цитохром С, APAF-1 претерпевает дальнейшее конформационное изменение, способствующее его олигомеризации и открывающее доступ CARD-домена APAF-1 для прокаспазы-9, которая тоже содержит CARD-домен. Так образуется конструкция, называемая тоже апоптосомой, с молекулярной массой > 1,3 млн дальтон, в составе которой - не менее 8 субъединиц APAF-1. Благодаря гомофильному CARD-CARD-взаимодействию с APAF-1 в эквимолярном соотношении связывается прокаспаза-9, а затем прокаспаза-9 связывает прокаспазу-3. Пространственное сближение молекул прокаспазы-9 на мультимерной арматуре из APAF-1-цитохром-С-комплексов, по-видимому, приводит к межмолекулярному протеолитическому процессингу прокаспазы-9 с образованием активной каспазы-9. Сходный механизм предложен для активации прокаспазы CED-3 у нематоды Caenorhabditis elegans - аналога прокаспазы-9 млекопитающих. Альтернативный вариант - прокаспаза-9, связавшись с апоптосомой, может принять конформацию, которая приводит к внутримолекулярному процессингу (самоактивации). Зрелая каспаза-9 затем расщепляет и активирует прокаспазу-3. Мутантный APAF-1, лишенный WD-40-повторов, активирует прокаспазу-9, но не способен к рекрутированию и активации прокаспазы-3. Флавопротеин AIF, будучи добавленным к изолированным ядрам из клеток HeLa, вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям печени крыс - высвобождение цитохрома С и каспазы-9. Микроинъекция AIF в интактные фибробласты крыс приводит к конденсации хроматина по переферии ядра, разрыву ДНК на крупные фрагменты длиной 50 т.п.н. и больше, снижению мембранного потенциала в митохондриях и переходу фосфатидилсерина из внутреннего слоя цитоплазматической мембраны в наружний. Ни один из этих эффектов AIF не предотвращается пептидным ингибитором каспаз N-бензоилоксикарбонил-Val-Ala-Asp.трифторметилкетоном (Z-VAD.fmk), который предотвращает апоптоз, индуцированный микроинъецированным цитохромом С. Эти данные показывают, что AIF является митохондриальным эффектором апоптоза у животных, действующим независимо от каспаз (Susin S. A. et al, 1999). Кроме рассмотренных компонентов, при нарушении наружной мембраны митохондрий из межмембранного объема выделяется термолабильный фактор, вызывающий необратимое превращение ксантиндегидрогеназы в ксантиноксидазу. Фактор устойчив к ряду испытанных ингибиторов протеаз, включая каспазы, сериновые и металлопротеазы. Ксантиндегидрогеназа катализирует зависимое от NAD+ окисление ксантина до гипоксантина и последующее окисление гипоксантина до мочевой кислоты. Ксантиноксидаза катализирует те же реакции, но не с NAD+, а с О2 в качестве акцептора электронов. При этом образуются О2A, Н2О2, а из них - и другие активные формы кислорода (АФК), которые разрушают митохондрии и являются мощными индукторами апоптоза. Механизмы образования АФК, конечно, не ограничиваются ксантиноксидазной реакцией. Главным источником АФК в клетках являются митохондрии. Резкое увеличение АФК происходит при возрастании мембранного потенциала в митохондриях, когда снижено потребление ATP и скорость дыхания лимитируется ADP. Доля электронного потока через дыхательную цепь митохондрий, идущая на образование О2A, достигает 1-5 %. Цитоплазматическая мембрана макрофагов и нейтрофилов, как уже отмечалось, содержит О2A - генерирующую NADPH-оксидазу. В зависимости от пути, по которому осуществляется активация каспаз, различают несколько типов клеток. Клетки типа I (в частности, линия лимфобластоидных В-клеток SKW и T-клетки линии Н9) подвергаются программированной гибели по пути, зависимому от апоптозных рецепторов плазматической мембраны без участия митохондриальных белков. Клетки типа II (например, линии Т-клеток Jurkat и СЕМ) погибают по пути апоптоза, зависимому от митохондриального цитохрома С. Запрограммированная клеточная смерть, вызванная химиотерапевтическими соединениями, УФ- или ?-облучением, по-видимому, напрямую связана с апоптозной функцией митохондрий: клетки, лишенные генов белка APAF-1 или каспазы-9, устойчивы к химио- и радиационной обработке, но погибают при индукции Fas-рецептора. Некоторые клетки, например, клетки эмбриональной нервной системы, включают механизмы апоптоза, если они испытывают дефицит апоптозподавляющих сигналов (называемых также факторами выживания) от других клеток. Физиологический смысл процесса - в элиминации избыточных нервных клеток, конкурирующих за ограниченное количество факторов выживания. Эпителиальные клетки при отделении от внеклеточного матрикса, вырабатывающего факторы выживания, тоже обречены на гибель. Факторы выживания связываются соответствующими цитоплазматическими рецепторами, активируя синтез подавляющих апоптоз агентов и блокируя стимуляторы апоптоза. Некоторые вещества (например, стероидные гормоны) оказывают дифференцированный эффект на различные типы клеток - предотвращают апоптоз одних типов клеток и индуцируют его у других. 5. Взаимодействие клеток с внеклеточным матриксом осуществляется с помощью интегринов. Интегрины - большое семейство гетеродимерных мембранных белков, которые участвуют в адгезии клеток, связывая внутриклеточный цитоскелет с лигандами внеклеточного матрикса. Нарушение адгезии клеток индуцирует апоптоз. Большинство интегринов специфически взаимодействует с трипептидным RGD (аргинин-глицин-аспартат)-мотивом, входящим в состав белков внеклеточного матрикса. Растворимые низкомолекулярные RGD-содержащие пептиды являются эффективными индукторами апоптоза: проникая в клетки, они активируют латентную каспазу-3. Ряд каспаз, включая каспазу-3, содержит RGD-последовательность вблизи активного центра фермента. В молекуле прокаспазы эта последовательность, вероятно, вовлечена во внутримолекулярное взаимодействие, придающее молекуле профермента такую конформацию, при которой протеазная активность не может проявиться. Предположительно RGD-последовательность взаимодействует с последовательностью DDM (аспартат-аспартат-метионин), локализованной вблизи участка протеолитической активации прокаспазы-3. Низкомолекулярный RGD-пептид, проникая в клетку и вступая в конкурентные взаимоотношения с RGD-последовательностью прокаспазы-3, вытесняет ее из сферы взаимодействия с DDM-последовательностью молекул профермента и индуцирует изменение их конформации, олигомеризацию и аутопроцессинг прокаспазы-3 с образованием активной каспазы-3.
2. Существует путь передачи сигнала апоптоза с участием эндоплазматического ретикулума (ЭР). В ЭР локализована прокаспаза-12. Нарушение внутриклеточного Ca2+-гомеостаза добавкой тапсигаргина или Ca2+-ионофорного антибиотика А23187 ведет к апоптозу клеток, вызванному превращением прокаспазы-12 в каспазу-12. ЭР-зависимый апоптоз связан с болезнью Альцгеймера: кортикальные нейроны мышей, дефицитных по каспазе-12, устойчивы к апоптозу, индуцированному І-амилоидным белком, но не к апоптозу с участием рецепторов плазматической мембраны или митохондриального цитохрома С. 3. Цитотоксические лимфоциты, Т-киллеры, могут вызывать апоптоз у инфицированных клеток с помощью белка перфорина. Полимеризуясь, перфорин образует в цитоплазматической мембране клетки-мишени трансмембранные каналы, по которым внутрь клетки поступают TNFb , гранзимы (фрагментины) - смесь сериновых протеаз. Существенным компонентом этой смеси является гранзим В - протеолитический фермент, превращающий прокаспазу-3 в активную каспазу-3. 4. Высвобождаемый из митохондрий цитохром С вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) участвует в активации каспазы-9. APAF-1 - белок с молекулярной массой 130 кДа, содержащий CARD-домен (caspase activation and recruitment domain) на N-конце и 12 повторяющихся аминокислотных WD-40-последовательностей (WD - дипептид из триптофана и аспартата) на С-конце, образует комплекс с прокаспазой-9 в присутствии цитохрома С и dATP или АТР. К наиболее охарактеризованным WD-белкам относится ?-cубъединица G-белков. Из этих субъединиц собираются жесткие, симметричные структуры, наподобие веера или пропеллера. WD-повторы свойственны белкам, участвующим в регуляции деления и дифференцировки эукариотических клеток, транскрипции генов, модификации мРНК, трансмембранной передачи сигналов, слияния мембранных везикул. Среди прокариот WD-белки обнаружены у цианобактерий. APAF-1 играет роль арматуры, на которой происходит аутокаталитический процессинг каспазы-9. Предполагается, что в результате зависимого от гидролиза dATP (или АТР) конформационного изменения APAF-1 приобретает способность связывать цитохром С. Связав цитохром С, APAF-1 претерпевает дальнейшее конформационное изменение, способствующее его олигомеризации и открывающее доступ CARD-домена APAF-1 для прокаспазы-9, которая тоже содержит CARD-домен. Так образуется конструкция, называемая тоже апоптосомой, с молекулярной массой > 1,3 млн дальтон, в составе которой - не менее 8 субъединиц APAF-1. Благодаря гомофильному CARD-CARD-взаимодействию с APAF-1 в эквимолярном соотношении связывается прокаспаза-9, а затем прокаспаза-9 связывает прокаспазу-3. Пространственное сближение молекул прокаспазы-9 на мультимерной арматуре из APAF-1-цитохром-С-комплексов, по-видимому, приводит к межмолекулярному протеолитическому процессингу прокаспазы-9 с образованием активной каспазы-9. Сходный механизм предложен для активации прокаспазы CED-3 у нематоды Caenorhabditis elegans - аналога прокаспазы-9 млекопитающих. Альтернативный вариант - прокаспаза-9, связавшись с апоптосомой, может принять конформацию, которая приводит к внутримолекулярному процессингу (самоактивации). Зрелая каспаза-9 затем расщепляет и активирует прокаспазу-3. Мутантный APAF-1, лишенный WD-40-повторов, активирует прокаспазу-9, но не способен к рекрутированию и активации прокаспазы-3. Флавопротеин AIF, будучи добавленным к изолированным ядрам из клеток HeLa, вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям печени крыс - высвобождение цитохрома С и каспазы-9. Микроинъекция AIF в интактные фибробласты крыс приводит к конденсации хроматина по переферии ядра, разрыву ДНК на крупные фрагменты длиной 50 т.п.н. и больше, снижению мембранного потенциала в митохондриях и переходу фосфатидилсерина из внутреннего слоя цитоплазматической мембраны в наружний. Ни один из этих эффектов AIF не предотвращается пептидным ингибитором каспаз N-бензоилоксикарбонил-Val-Ala-Asp.трифторметилкетоном (Z-VAD.fmk), который предотвращает апоптоз, индуцированный микроинъецированным цитохромом С. Эти данные показывают, что AIF является митохондриальным эффектором апоптоза у животных, действующим независимо от каспаз (Susin S. A. et al, 1999). Кроме рассмотренных компонентов, при нарушении наружной мембраны митохондрий из межмембранного объема выделяется термолабильный фактор, вызывающий необратимое превращение ксантиндегидрогеназы в ксантиноксидазу. Фактор устойчив к ряду испытанных ингибиторов протеаз, включая каспазы, сериновые и металлопротеазы. Ксантиндегидрогеназа катализирует зависимое от NAD+ окисление ксантина до гипоксантина и последующее окисление гипоксантина до мочевой кислоты. Ксантиноксидаза катализирует те же реакции, но не с NAD+, а с О2 в качестве акцептора электронов. При этом образуются О2A, Н2О2, а из них - и другие активные формы кислорода (АФК), которые разрушают митохондрии и являются мощными индукторами апоптоза. Механизмы образования АФК, конечно, не ограничиваются ксантиноксидазной реакцией. Главным источником АФК в клетках являются митохондрии. Резкое увеличение АФК происходит при возрастании мембранного потенциала в митохондриях, когда снижено потребление ATP и скорость дыхания лимитируется ADP. Доля электронного потока через дыхательную цепь митохондрий, идущая на образование О2A, достигает 1-5 %. Цитоплазматическая мембрана макрофагов и нейтрофилов, как уже отмечалось, содержит О2A - генерирующую NADPH-оксидазу. В зависимости от пути, по которому осуществляется активация каспаз, различают несколько типов клеток. Клетки типа I (в частности, линия лимфобластоидных В-клеток SKW и T-клетки линии Н9) подвергаются программированной гибели по пути, зависимому от апоптозных рецепторов плазматической мембраны без участия митохондриальных белков. Клетки типа II (например, линии Т-клеток Jurkat и СЕМ) погибают по пути апоптоза, зависимому от митохондриального цитохрома С. Запрограммированная клеточная смерть, вызванная химиотерапевтическими соединениями, УФ- или ?-облучением, по-видимому, напрямую связана с апоптозной функцией митохондрий: клетки, лишенные генов белка APAF-1 или каспазы-9, устойчивы к химио- и радиационной обработке, но погибают при индукции Fas-рецептора. Некоторые клетки, например, клетки эмбриональной нервной системы, включают механизмы апоптоза, если они испытывают дефицит апоптозподавляющих сигналов (называемых также факторами выживания) от других клеток. Физиологический смысл процесса - в элиминации избыточных нервных клеток, конкурирующих за ограниченное количество факторов выживания. Эпителиальные клетки при отделении от внеклеточного матрикса, вырабатывающего факторы выживания, тоже обречены на гибель. Факторы выживания связываются соответствующими цитоплазматическими рецепторами, активируя синтез подавляющих апоптоз агентов и блокируя стимуляторы апоптоза. Некоторые вещества (например, стероидные гормоны) оказывают дифференцированный эффект на различные типы клеток - предотвращают апоптоз одних типов клеток и индуцируют его у других. 5. Взаимодействие клеток с внеклеточным матриксом осуществляется с помощью интегринов. Интегрины - большое семейство гетеродимерных мембранных белков, которые участвуют в адгезии клеток, связывая внутриклеточный цитоскелет с лигандами внеклеточного матрикса. Нарушение адгезии клеток индуцирует апоптоз. Большинство интегринов специфически взаимодействует с трипептидным RGD (аргинин-глицин-аспартат)-мотивом, входящим в состав белков внеклеточного матрикса. Растворимые низкомолекулярные RGD-содержащие пептиды являются эффективными индукторами апоптоза: проникая в клетки, они активируют латентную каспазу-3. Ряд каспаз, включая каспазу-3, содержит RGD-последовательность вблизи активного центра фермента. В молекуле прокаспазы эта последовательность, вероятно, вовлечена во внутримолекулярное взаимодействие, придающее молекуле профермента такую конформацию, при которой протеазная активность не может проявиться. Предположительно RGD-последовательность взаимодействует с последовательностью DDM (аспартат-аспартат-метионин), локализованной вблизи участка протеолитической активации прокаспазы-3. Низкомолекулярный RGD-пептид, проникая в клетку и вступая в конкурентные взаимоотношения с RGD-последовательностью прокаспазы-3, вытесняет ее из сферы взаимодействия с DDM-последовательностью молекул профермента и индуцирует изменение их конформации, олигомеризацию и аутопроцессинг прокаспазы-3 с образованием активной каспазы-3.
Характеристика белков Вcl-2
Белки семейства Bcl-2 играют центральную роль в выборе между жизнью и смертью клетки. Bcl-2 гомолог белка CED-9 у Caenorhabditis еlegans, первоначально был открыт как протоонкоген, обнаруженный в результате хромосомной транслокации t(14;18) в случае В-клеточной лимфомы. У Bcl-2 первого была обнаружена способность предотвращать апоптоз, индуцированный отсутствием итерлейкина-3 в культуре В-лимфоцитов человека. С тех пор были обнаружены многочисленные гомологи Bcl-2. Это семейство структурно сходных белков включает более двух десятков членов, в том числе продукты протоонкогенов Bcl-2 и Bcl-x, обладающие способностью блокировать апоптоз, и опухолевый супрессор Bax, наоборот, индуцирующий апоптоз. Семейство Bcl-2 белков можно разделить на три основные группы: 1. Антиапоптогенные молекулы, такие как Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1(Bfl-1) и Boo. Все они обладают противоапоптозной активностью и имеют четыре группы гомологичных последовательностей BH1, ВН2, ВН3 и ВН4 домены, хотя у некоторых из них ВН4 домен отсутствует. 2. Проапоптогенные молекулы Bax, Bak, Bad, Mtd(Bok) и Diva имеют гомологичные последовательности BH1, ВН2и ВН3, а ВН4 домен у них отсутствует. 3. Проапоптогенные белки, содержащие только ВН3 домен: Bik, Bid, Bim, Hrk(DP5), Blk и Bnip3, Bnip3L. Было показано, что ВН1-3 домены играют важную роль в формировании гетеро- и гомо-димеров между проапоптогенными и антиапоптогенными членами семейства, и клеточные уровни этих димеров могут сыграть определяющую роль в судьбе клетки. Гетеродимеризация происходит посредством взаимодействия BH-3 домена проапоптогенного белка с гидрофобным комплексом, образованным BH-1, BH-2 и BH-3 доменами антиапоптогенных белков. Важно то, что домены BH-1 и BH-2, BH-4 необходимы для антиапоптогенной активности белка, в то время как BH-3 домен необходим и достаточен для проапоптогенной активности. Белок Bcl-2 один может связывать, по крайней мере, пять членов семейства, и эта его функция может быть, кроме того, дополнена возможностью посттрансляционной модификации с помощью фосфорилирования. Близкий ген, bcl-x, кодирует два белка, различающихся сплайсингом РНК, Bcl-xL и Bcl-xS. Также как Bcl-2, белок Bcl-xL ингибирует апоптоз, в то время как белок Bcl-xS оказывает негативный эффект на функцию Bcl-2 и Bcl-xL. Повышенная экспрессия генов этих белков может приводить к устойчивости к большинству вызывающим апоптоз стимулам, так как к этим белкам сходится множество путей апоптоза. Большинство антиапоптогенных членов семейства Bcl-2 содержат на С-концевом участке гидрофобную последовательность, которая необходима для связывания с внутриклеточными мембранами. Таким образом, проапоптогенные и антиапоптогенные члены семейства Bcl-2, в отсутствие сигналов смерти, локализованы в различных внутриклеточных компартментах. Антиапоптогенные молекулы представляют собой мембранные белки, которые находятся в митохондрии, эндоплазматическом ретикулуме и в ядерной мембране. Проапоптогенные молекулы семейства Bcl-2 в основном локализованы в цитозоле или связаны с цитоскелетом. Механизм, с помощью которого белки семейства Bcl-2 регулируют митохондриально-зависимый апоптоз, остается спорным. Недавно было установлено, что VDAC (Voltage-dependent anion channel) является одним из функциональных мишеней этих белков. Белки семейства Bcl-2, такие как Bax, Bak, Bcl-2 и Bcl-XL могут взаимодействовать с двумя компонентами РТ (permeability transition) пор, с VDAC, локализованным на наружной митохондриальной мембране, и с ANT (adenine nucleotide translocator), на внутренней мембране. Bcl-2 и Bcl-XL закрывают VDAC-каналы, через которые осуществляется выброс цитохрома С и/или AIF. Bax и Bak, находящиеся в норме в определенных компартментах цитоплазмы, при апоптогенных сигналах перемещаются в митохондриальные мембраны, где они, взаимодействуя с интегральным белком наружной митохондриальной мембраны VDAC, стимулируют открытие канала. Кроме того, Bax образует гетеромерные комлексы с белками Bcl-2, Bcl-x, что, возможно, открывает закрытые до этого каналы. Таким образом, баланс между про- и антиапоптогенными членами семейства Bcl-2 играет решающую роль в судьбе клетки.Заключение
К настоящему времени известно несколько миллионов химических соединений как природного, так и антропогенного происхождения. Многие из них являются токсическими для животных и человека. Большинство токсических веществ подвергаются метаболизму, который осуществляется, главным образом, в печени. Ключевую роль в метаболизме ксенобиотиков играют ферменты 1-й фазы (цитохром Р450) и 2-фазы (трансферазы). Однако наряду с реакциями детоксификации в клетке могут осуществляться реакции токсификации. Этот процесс связан с образованием высоко активных "реактивных" метаболитов, способных связываться с нуклеофильными сайтами макромолекул, в том числе и с ДНК. Существуют системы защиты от ДНК-повреждающих агентов. Идентифицировно множество систем репарации ДНК, начиная с бактерий и заканчивая человеком. Дефекты репарации, нарушение репликации ДНК могут привести к возникновению и фиксации мутаций. Особенно опасным является ситуация, когда мутация произошла генах-мишенях, регулирующих такие жизненно важные процессы, как клеточная дифференцировка, клеточное деление, программированная клеточная гибель. Наше понимание основных механизмов действия токсических соединений на живые системы поможет нам не только понять причины возникновения многих болезней (в том числе и рака), но и научиться бороться с ними.Библиографический список
1. Гуляева Л.Ф., Гришанова А.Ю. и др. Микросомная монооксигеназная система живых организмов в биомониторинге окружающей среды. Аналитический обзор. ГПНТБ, Новосибирск, 1994 - 98 с. 2. Гуляева Л.Ф., Вавилин В.А., Ляхович В.В. Ферменты биотрансформации ксенобиотиков в химическом канцерогенезе. Аналитический обзор. ГПНТБ, Новосибирск, 2000 - 90 с. 3. Claassen C.D. " Toxicology . The basic Science of poisons". l - New York, Chicago, Toronto, London. Sixth Edition, 2001 - 1236p. 4. McKinnell R.G., Parchment R.E., Perantoni A.O., Pierce G.B. "The Biological Basis of Cancer", Cambridge University Press, 1998 - 378p. 5. David P. Josephy. "Molecular Toxicology", Oxford University Press, 1997 - 367 p. 6. Lewin "Gene", Oxford University Press, 2000 - 990p. 7. В.Д. Самуилов, А.В. Алескин, Е.М., Лагунова. Программированная клеточная гибель. Обзор. Биохимия, 2000, т. 65, вып. 8, с. 1029-1046. 8. Обзорные статьи по биохимии рака. Биохимия, 2000, т. 65, вып. 1, с. 3 -139 9. Robert H. Rice. Biological effects of toxoc compounds. Syllabus. University of California, Davis, 2002. - p. 150
Дата: 2019-02-02, просмотров: 795.