Методические указания для проведения лабораторных занятий
по курсу «Биохимия сельскохозяйственной продукции»
(для бакалавров технологического факультета по направлению: 35.03.07 –
Технология производства и переработки сельскохозяйственной
продукции)
Тверь, 2017
Составитель: к. с.-х. н., доцент кафедры агрохимии и земледелия
Шилова О.В.
Рецензент: к. б. н., доцент кафедры биологии животных, зоотехнии и основ
ветеринарии Тверской ГСХА Корецкая Е.А.
Шилова О.В. Методические указания для проведения лабораторных занятий по курсу «Биохимия сельскохозяйственной продукции» (для бакалавров технологического факультета по направлению: 35.03.07 – Технология производства и переработки сельскохозяйственной продукции) – Тверь, 2017– 69 с.
Председатель методической комиссии  Акимов А.А.
Акимов А.А.
Введение
Цели дисциплины: формирование современных представлений, знаний и умений о превращениях веществ и энергии в живых организмах, химическом составе сельскохозяйственной продукции растительного и животного происхождения, биохимических процессах, происходящих в ней при хранении и переработке.
Задачи дисциплины: изучение строения и биологических функций важнейших органических веществ; механизмов ферментативных и биоэнергетических превращений в организмах; химического состава сельскохозяйственной продукции и биохимических процессов, происходящих в ней при хранении и переработке;
– оценка качества и технологических свойств сельскохозяйственной продукции по биохимическим показателям;
– применение знаний о химическом составе и биохимических процессах при обосновании технологий производства, хранения и переработки сельскохозяйственной продукции;
– ознакомление с современными методами и достижениями биохимической науки.
Ход работы
1. Извлечение сахаров водой
Взвесить 5 г исследуемого материала (яблоки, капуста, морковь, лук), размельчить и растереть до гомогенной массы в фарфоровой ступке с небольшим количеством воды, нагретой до 70оС. Растертую массу количественно перенести в коническую колбу на 100 мл, объем вытяжки довести до 50 мл горячей дистиллированной водой и оставить на 10 мин для экстракции. По истечении указанного времени экстракт охладить, отфильтровать через воронку Шотта и количественно перенести вытяжку в мерную колбу. При необходимости довести объем водой до 50 мл.
Обязательно провести осветление вытяжки в случае окрашенных (мутных) экстрактов. Для этого в теплую, не доведенную до окончательного объема вытяжку добавить по каплям 10 % раствор уксуснокислого свинца до прекращения образования осадка. Обычно на вытяжку, полученную из навески 10 г, необходимо добавить 0,5-2 мл раствора уксуснокислого свинца. Избытка соли свинца следует избегать, так как он проходит через фильтр и мешает ходу анализа. Осветленную вытяжку отфильтровать и довести объем водой до 50 мл.
2. Определение восстанавливающих сахаров
Для определения содержания восстанавливающих сахаров предварительно следует приготовить раствор глицерата меди. Для этого необходимо сделать смесь (2:1) растворов № 1 (0,8 % CuSO4 · 5H2O) и № 2 (15 % NaOH с добавлением 1 мл глицерина).
Отобрать 1 мл осветленной отфильтрованной вытяжки в пробирку, добавить 15 мл глицерата меди, перемешать и нагревать на водяной бане при
70оС 6 мин. Затем пробирку охладить в холодной воде и отобрать прозрачную жидкость в кювету для определения оптической плотности раствора (проба 1).
3. Определение суммы восстанавливающих и невосстанавливающих сахаров (сахарозы)
Для определения содержания невосстанавливающих сахаров отобрать 0,5 мл осветленной отфильтрованной вытяжки, добавить 0,5 мл 1 % НСl, перемешать и поставить на кипящую водяную баню на 15 мин. По истечении указанного времени добавить 15 мл глицерата меди и нагревать на водяной бане ровно 6 мин. Охладить пробирку в холодной воде, дать вытяжке отстояться и отобрать прозрачную жидкость для оптического анализа (проба 2). В этой пробе определяется сумма восстанавливающих сахаров и сахарозы.
4. Спектрофотометрия
Оптическую плотность исследуемых растворов необходимо регистрировать на спектрофотометре при длине волны λ = 582 нм.
Содержание сахаров в пробах можно определить по калибровочной кривой, построенной по глюкозе. Для этого необходимо приготовить 50 мл раствора, содержащего 10 мг/мл глюкозы, и затем методом разбавления получить остальные растворы согласно таблице:
| № | Содержание глюкозы, мг/мл | Количество ис- ходного раствора глюкозы, мл | Количество воды, мл | Оптическая плотность, D582 |
| 1 2 3 4 5 6 | 0,5 1,0 2,5 5,0 7,5 10,0 | 0,5 1,0 2,5 5,0 7,5 10,0 | 9,5 9,0 7,5 5,0 2,5 0 |
Для определения оптической плотности ( D582) этих растворов предварительно провести реакции с глицератом меди, аналогично опытным образцам.
Количество восстанавливающих сахаров в исследуемом объекте (А, %) вычислить по формуле:
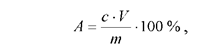
где с - содержание сахаров в пробе 1, найденное по калибровочной кривой; V - объем вытяжки, полученной из навески; m - масса навески в граммах.
По этой же формуле рассчитать сумму восстанавливающих сахаров и сахарозы, подставляя вместо с содержание сахаров в пробе 2, найденное по калибровочной кривой. Разность между вторым и первым определениями, умноженная на коэффициент 0,95, дает содержание сахарозы в исследуемом объекте.
Результаты оформить в таблицу.
| Объект | D582 проба 1 | D582 проба 2 | Количество вос- станавливающих сахаров, мг/г | Сумма моно- и олигосахаридов, мг/г | Количество сахарозы, мг/г |
Сделать выводы.
Контрольные вопросы
1. Классификация углеводов.
2. Основные моносахариды растений, их свойства и функции.
3. Основные дисахариды растений – сахароза, мальтоза, целлобиоза. Функции сахарозы в растениях.
4. Редуцирующие веществами – особенности строения и обнаружения их в растительном материале. Приведите примеры.
5. Нередуцирующие веществами – особенности строения и обнаружения их в растительном материале. Приведите примеры.
6. Использование растительных углеводов в пищевой промышленности.
Тема 2. Состав, строение и биологические функции углеводов и липидов
Лабораторная работа № 3. Определение кислотного и йодного числа
растительных жиров
Учебные вопросы:
1.Строение и химические свойства липидов
2.Физиологическая роль липидов для растений
3.Обмен липидов в растениях
Запасные жиры растений представляют собой сложные эфиры многоатомных спиртов (в первую очередь, глицерина) и жирных кислот – триглицериды. Этерификация глицерина может осуществляться как одним типом жирных кислот, так и разными, причем последний вариант наиболее распространен. В том случае, когда все три кислоты насыщенные, образуются твердые жиры, если кислоты ненасыщенные – жидкие. Растительные жиры чаще всего жидкие, поэтому их именуют маслами.
Для жиров (как жидких, так и твердых) характерна высокая степень гидрофобности. Они не растворимы в воде, но хорошо растворимы в углеводородах, галагеналканах, спиртах, эфирах.
В числе компонентов, входящих в состав растительных масел, всегда присутствует небольшое количество свободных жирных кислот. Этот показатель именуется кислотным числом и дает представление о количестве свободных жирных кислот в масле. В частности, кислотное число показывает, сколько мг КОН необходимо для нейтрализации свободных жирных кислот в 1 г масла.
Поскольку при длительном хранении масла происходят процессы окисления, то содержание свободных жирных кислот со временем увеличивается. Следовательно, кислотное число является важным показателем качества жира.
Другим физико-химическим показателем, характеризующим свойства жира, является число омыления, которое показывает количество мг КОН, необходимое для омыления связанных и нейтрализации свободных жирных кислот в 1 г масла.
О содержании ненасыщенных жирных кислот в жире судят по йодному числу – количеству граммов йода, присоединившемуся к 100 граммам жира. Определение физико-химических показателей растительного масла имеет важное значение, так как позволяет контролировать качество получаемого масличного сырья и его изменение при переработке и хранении.
Цель настоящей работы – определить и сравнить физико-химические свойства различных растительных масел.
Реактивы и материалы: подсолнечное, рапсовое, оливковое масло, этиловый спирт, КОН, H2SO4, фенолфталеин, хлороформ, 2,5 % спиртовой раствор иода, гипосульфит натрия, крахмал, дистиллированная вода, конические колбы, весы, микробюретка для титрования, водяная баня, обратный холодильник.
Ход работы
1. Определение кислотного числа
Взвесить пустую колбу, затем осторожно туда добавить небольшое количество растительного масла (оливковое, подсолнечное, рапсовое). Массу навески определить по разности массы колбы с маслом и пустой колбы.
Приготовить 100 мл 0,2 н спиртового раствора КОН. Для этого взвесить 1,12 г щелочи и растворить в минимальном количестве воды (1-1,5 мл). После того как навеска щелочи растворена, довести объем 96 % этанолом до 100 мл.
В колбу с маслом прилить 15 мл этанола, добавить 2-3 капли фенолфталеина и тщательно перемешать. Затем оттитровать полученный раствор 0,2 н спиртовым раствором щелочи до появления окрашивания. Титрование более слабыми растворами КОН не дает видимых изменений окраски и затрудняет определение конца титрования. В качестве контроля оттитровать 15 мл этанола с 2-3 каплями фенолфталеина тем же раствором щелочи.
Кислотное число (КЧ) вычислить по формуле:
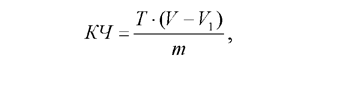
где T - титр щелочи (количество КОН в 1 мл раствора), мг; V - объем раствора щелочи, пошедшей на титрование контрольной пробы, мл; VI - объем раствора щелочи, пошедшей на титрование опытной
пробы, мл; m - масса навески в граммах.
2. Определение числа омыления
В конической колбе взвесить навеску масла 0,2-0,5 г (как указано выше), прилить 15 мл 0,2 н спиртового раствора щелочи и нагревать на водяной бане 30 мин с обратным холодильником. Одновременно прокипятить контрольную пробу с таким же количеством щелочи, но не содержащую масла. По истечении указанного времени охладить колбы с раствором. При полном омылении на опытной колбе не должно быть блестящих капелек масла, в противном случае омыление надо продолжить. После омыления и охлаждения в колбы добавить 2-3 капли фенолфталеина и избыток щелочи оттитровать 0,5 . раствором H2SO4, пользуясь микробюреткой.
Вычислить число омыления (ЧО) по формуле:
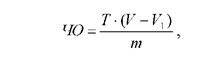
где T - титр щелочи, мг; V - объем кислоты, пошедшей на титрование контрольной пробы, мл; V1 - объем кислоты, пошедшей на титрование опытной пробы, мл; m - масса навески в граммах.
3. Определение йодного числа
В коническую колбу взвесить 0,1 грамм жира и внести 5 мл хлороформа. Для контроля взять колбу, содержащую только 5 мл хлороформа. В обе колбы прилить по 10 мл 2,5 % спиртового раствора иода, закрыть пробкой, тщательно перемешать и оставить в темной месте на 1 ч при комнатной температуре. По истечении указанного времени избыток иода оттитровать 0,1 н раствором гипосульфита натрия до желтой окраски. Затем добавить 1 мл 1 % раствора крахмала и продолжить титрование до обесцвечивания раствора. Иодное число (ИЧ) вычислить по формуле:
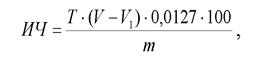
где T - титр 0,1 н раствора гипосульфита натрия, мг; V - объем раствора гипосульфита, пошедшего на титрование контрольной пробы, мл; V 1 - объем раствора гипосульфита натрия, пошедшей на титрование опытной пробы, мл; m - масса навески в граммах; 0,0127 - количество йода (г), эквивалентное 1 мл 0,1 н раствора гипосульфита натрия.
Результаты оформить в виде таблицы.
| Вид растительного масла | Кислотное число | Число омыления | Йодное число |
Сделать сравнительный анализ полученных результатов.
4.Определение перекисного числа
В присутствии кислорода воздуха кислоты, входящие в состав жиров, могут частично окисляться и образовывать перекиси. Это явление наблюдается при порче жиров, а также при их высыхании. Таким образом, перекис-ное число служит показателем окислительных изменений жиров.
Обычно перекисное число, как и йодное число, выражается в граммах йода, которое может прореагировать с активным водородом перекисей, содержащихся в 100 г жира.
Принцип метода. Определение перекисного числа основано на том, что перекиси жира в кислой среде способны реагировать с йодистым калием, выделяя из него йод.
Выделяющийся йод оттитровывают раствором гипосульфита:
2Na2S2О3 + J2 → 2NaJ + Na2S4О6
и по количеству гипосульфита, затраченному на связывание выделившегося йода, вычисляют перекисное число.
Ход работы
1. На аналитических весах отвешивают 2 –3 г жира, который помещают в коническую колбу емкостью 150–200 мл. В другую колбу (контроль) наливают 2–3 мл воды.
2. В обе колбы наливают по 10 мл хлороформа и растворяют жир.
3. В колбы добавляют по 20 мл ледяной уксусной кислоты и по 1 мл свежеприготовленного насыщенного раствора йодистого калия.
4. Смесь тщательно перемешивают и оставляют на 3 минуты.
5. Проводят титрование выделившегося йода 0,01 н. раствором гипосульфита. Титруют до появления желтой окраски.
6. Затем в колбы прибавляют по 1 мл 1%-ного раствора крахмала и титруют до исчезновения голубой окраски.
Вычисление результатов можно проводить по следующей формуле:
х = _(а - б) · Т · 0,001269 ·100
Н
где х – перекисное число;
а – количество миллилитров 0,01 н. гипосульфита, израсходованное на титрование йода, выделившегося в результате реакции жира с йодистым калием;
б – количество миллилитров 0,01 н. гипосульфита, израсходованное в контрольном определении;
Т – поправка к титру раствора гипосульфита;
Н – навеска жира.
Контрольные вопросы
1. Строение, свойства и функции липидов в живой клетке.
2. Жирные кислоты в составе липидов, их влияние на качество пищевых
продуктов.
3. Особенности обмена липидов растений.
4. Глиоксилатный цикл.
5. Содержание жиров в семенах и плодах культурных растений.
6. Свойства основных растительных масел.
7. Сравнительный анализ растительных и животных жиров.
8. Использование липидов в пищевой промышленности?
Ход работы
1. Выделение суммарных белков из семян зерновых и зернобобовых растений
Навеску 1 г сырых проросших семян (пшеница, ячмень, фасоль, горох, люпин, соя) поместить в фарфоровую ступку. Добавить небольшое количество кварцевого песка и тщательно растереть с 40 мл фосфатного буфера (рН 8), содержащего 0,2 % бисульфита натрия и несколько капель этилового спирта. При использовании сухих семян их необходимо предварительно размолоть до состояния муки, и после добавления фосфатного буфера настоять в течение часа.
Для приготовления 0,1 М фосфатного буфера с рН 8,0 необходимо взять 95 мл 0,2 М раствора Na2HPO4 · 2H2O добавить 5 мл 0,2 М раствора NaH2PO4 · H2O и довести объем водой до 200 мл.
Гомогенат количественно перенести в коническую колбу на 100 мл и довести объем фосфатным буфером до 50 мл. Содержимое колб перемешивать на ротаторе в течение 1 ч. По истечении указанного времени колбы достать из ротатора и дать вытяжке отстояться в течение 15 мин. Затем при помощи пипетки из верхней части гомогената осторожно отобрать 10 мл раствора и перенести в центрифужные пробирки. Провести центрифугирование раствора в течение 15 мин при 3000 об/мин при комнатной температуре. По окончании центрифугирования из пробирок отобрать пипеткой 1 мл надосадочной жидкости и перенести в другую пробирку. Туда же необходимо добавить 9 мл фосфатного буфера с рН 8, тщательно перемешать, после чего раствор суммарных белков готов к спектрофотометрированию.
2. Спектрофотометрический метод определения суммарных белков
Раствор белков налить в кварцевую кювету и определить его оптическую плотность при 280 и 260 нм (в качестве раствора сравнения использовать исходный раствор фосфатного буфера). Ароматические аминокислоты тирозин и триптофан, содержащиеся в белках, поглощают свет в области λ = 280 нм. Однако поглощение в этой области имеют и нуклеиновые кислоты, хотя максимум поглощения последних составляет λ = 260 нм. Поэтому для определения концентрации белка в растворах названным методом используют уравнение Варбурга - Христиана:
с = 1,55 × D 280 - 0,76 × D 260
где с - содержание белка в мг/мл; D 280 и D260 - оптическая плотность растворов при 280 и 260 нм; 1,55 и 0,76 - расчетные коэффициенты.
Содержание суммарных белков в навеске семян (мг/г сухой массы) рассчитать по формуле:

где c - содержание белка в растворе, найденное по формуле Варбурга - Христиана; V - объем вытяжки в мл; m - масса навески в граммах;
10 - коэффициент, учитывающий разведение вытяжки; 86 - сухая масса воздушно-сухой массы навески семян, %; 60 - сухая масса проросших семян, %.
Результаты оформить в виде таблицы.
| Культура | Масса навески, г | Объем вытяжки, мл | Оптическая плотность | Концентрация белка в пробе, мг/мл | Концентра- ция белка в растении, мг/г | |
| D 280 | D260 | |||||
Сделать сравнительный анализ полученных результатов.
3. Выделение белков из вегетативных органов растений
Взвесить навеску 0,5 г свежего растительного материала (листья ячменя, клевера, клубни картофеля), измельчить и растереть в фарфоровой ступке с 4-кратным (по массе) количеством фосфатного буфера с рН 8, содержащим 0,2 % бисульфида натрия и несколько капель этилового спирта. Гомогенат залить 10 мл холодной 5 % ТХУ, тщательно перемешать и поместить в холодильник на 20 мин. Затем гомогенат перенести в центрифужные пробирки и центрифугировать в течение 15 мин при 6000 об/мин для осаждения белков. После центрифугирования надосадочную жидкость слить. Осадок промыть охлажденным 96 % этанолом до полного исчезновения зеленой окраски в надоса-дочной жидкости. Повторить центрифугирование и к промытому осадку добавить 2 мл 0,5 н NaOH. Поместить пробирки на 5 мин на кипящую водяную баню, после чего прилить еще 5 мл 0,5 н NaOH, перемешать и центрифугировать 10 мин при 6000 об/мин. Надосадочную жидкость перенести в мерный цилиндр и измерить общий объем щелочного гидролизата. Количество белка в пробе определить по методу Лоури.
4. Определение белков по методу Лоури
Для определения содержания белков по методу Лоури необходимо предварительно приготовить растворы:
А - 2 % Na2CO3 в 0,1 н NaOH;
B - 0,5 % CuSO4 · 5H2O в 1 % K-Na-виннокислом;
C - смесь растворов А и В (50:1 по объему);
Е - реактив Фолина.
Отобрать 1 мл щелочного гидролизата, добавить 5 мл раствора С, перемешать и через 10 мин добавить 0,5 мл раствора Е. Смесь выдержать в течение 30 мин при комнатной температуре, после чего измерить оптическую плотность раствора при 750 нм на спектрофотометре. В качестве раствора сравнения использовать смесь: 1 мл 0,5 н NaOH, 5 мл раствора С и 0,5 мл реактива Фолина.
Для определения содержания белка в растворе необходимо построить калибровочный график по альбумину или казеину.
На аналитических весах взвесить 100 мг альбумина и растворить в 100 мл фосфатного буфера. Из этого стандартного раствора приготовить производные растворы с содержанием белка от 0,05 до 1,0 мг/мл согласно таблице:
| № | Содержание белка, мг/мл | Стандартный раствор альбумина, мл | Фосфатный буфер, мл | Оптическая плотность, D750 |
| 1 | 0,05 | 0,5 | 9,5 | |
| 2 | 0,1 | 1,0 | 9,0 | |
| 3 | 0,2 | 2,0 | 8,0 | |
| 4 | 0,3 | 3,0 | 7,0 | |
| 5 | 0,4 | 4,0 | 6,0 | |
| 6 | 0,5 | 5,0 | 5,0 | |
| 7 | 1,0 | 10 | 0 |
Последовательно отобрать 1 мл каждого из растворов альбумина, перенести в чистые предварительно пронумерованные пробирки и добавить, как и в случае опытных растворов, 5 мл раствора С, а через 10 мин 0,5 мл раствора Е. Содержимое каждой из пробирок осторожно перемешать и оставить при комнатной температуре на 30 мин. По истечении указанного времени измерить оптическую плотность растворов при длине волны λ = 750 нм.
По данным, полученным для стандартных растворов, построить калибровочный график (на оси ординат – величина оптической плотности, на оси абсцисс – концентрация белка).
Пользуясь калибровочной кривой найти концентрацию белка в исследуемом растворе. Общее содержание белка в вегетативной части растения (мг/г сырой массы) рассчитать с учетом где с – концентрация белка в исследуемом растворе в мг/мл; V – объем щелочного гидролизата в мл; m – масса навески растительного материала в граммах.
Результаты оформить в виде таблицы.
| Культура | Масса навески, г | Оптическая плотность, D750 | Содержание белка в растворе, мг/мл | Содержание белка в растении, мг/г |
Сделать выводы.
Контрольные вопросы
1. Протеиногенные и непротеиногенные аминокислоты.
2. Биосинтез и функции непротеиногенных аминокислот.
3. Особенности строения белков и их физиологическая роль в живой клетке.
4. Белки семян и листьев растений.
5. Особенности белкового состава зерновых и зернобобовых культур.
6. Способы обнаружения белка в неизвестном объекте.
7. Проблемы, связанные с изучением растительных белков.
Ход работы
Для количественного определения восстановленной аскорбиновой кислоты взять навеску свежего растительного материала массой 5 г, измельчить и перенести в фарфоровую ступку. Залить сырье 10 мл 2 % метафосфорной или 1 % щавелевой кислоты и быстро растереть. Гомогенат количественно перенести в мерную колбу на 100 мл, объем довести до метки той же кислотой. Содержимое колбы тщательно перемешать, после чего профильтровать или отцентрифугировать 10 мин при 1000 об/мин.
Из фильтрата взять три пробы по 10 мл и перенести в чистые пробирки. К каждой пробе добавить 1 мл свежеприготовленного 0,025 % раствора 2,6-дихлорфенолиндофенола. Для приготовления этого раствора необходимо взять 0,0625 г 2,6-дихлорфенолиндофенола и растворить в 250 мл теплой (45 °С) дистиллированной воды, добавить 6 капель 0,01 н. щелочи.
Если при добавлении 1 мл 2,6-дихлорфенолиндофенола растительная вытяжка полностью обесцвечивается (это наблюдается при значительном количестве аскорбиновой кислоты в исследуемом объекте), то ее нужно разбавить в 2 раза метафосфорной кислотой.
После добавления 2,6-дихлорфенолиндофенола к растительному экстракту включить секундомер, содержимое пробирки тщательно перемешать и быстро залить раствор в кювету толщиной 10 мм.
Через 35 секунд определить оптическую плотность раствора на спектрофотометре при длине волны λ = 530 нм. В качестве раствора сравнения использовать смесь метафосфорной кислоты и 2,6-дихлор-фенолиндофенола (10:1).
Изменение в интенсивности окрашивания опытного образца про-порционально количеству аскорбиновой кислоты, находящейся в рас-тительной вытяжке. Количественный расчет произвести по калибровочной кривой.
Для построения калибровочной кривой необходимо: приготовить стандартный раствор аскорбиновой кислоты концентрацией 1 мг/мл, для этого взвесить 100 мг аскорбиновой кислоты и растворить в 100 мл 2 % метафосфорной кислоты;
– из стандартного раствора приготовить разведения, содержащие от 1 до 100 мкг/мл аскорбиновой кислоты, согласно таблице;
– отобрать 10 мл каждого из растворов аскорбиновой кислоты, перенести в чистые предварительно пронумерованные пробирки, добавить 1 мл 2,6-ди
хлорфенолиндофенола и определить оптическую плотность этих растворов.
| № | Содержание аскорбиновой кислоты, мкг/мл | Количество стандартного раствора, мл | Количество воды, мл | Оптическая плотность раствора, D530 |
| 1 | 1 | 1 | 999 | |
| 2 | 5 | 1 | 199 | |
| 3 | 10 | 1 | 99 | |
| 4 | 20 | 2 | 98 | |
| 5 | 50 | 5 | 95 | |
| 6 | 100 | 10 | 90 |
Пользуясь калибровочной кривой, найти концентрацию аскорбиновой кислоты в вытяжке. Количество аскорбиновой кислоты в исследуемом материале (мгк/г сырой массы) вычислить по формуле:
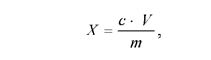
где с – содержание аскорбиновой кислоты в 1 мл вытяжки, найденное по калибровочной кривой; V – объем экстракта в мл; m – масса исследуемого материала в граммах.
Результаты представить в виде таблицы.
| Объект исследования | Масса навески, г | Оптическая плотность раствора, D530 | Количество аскорбиновой кислоты в вытяжке, мкг/мл | Количество аскорбиновой кислоты в объекте, мкг/г |
Сделать анализ полученных результатов.
Определение витаминов В1 и В2 в растениях
В растениях содержание витаминов невелико. Вместе с тем отдельные органы и ткани растений способны в большей степени накапливать их в своем составе. Так, овощные и плодовые культуры характеризуются высоким содержанием каротиноидов, витамина Р, аскорбиновой и фолиевой кислотой. В семенах злаковых и зернобобовых культур много рибофлавина, тиамина, ниацина. В зеленых листьях растений накапливаются филлохиноны.
Тиамин, или витамин В1, в виде тиаминпирофосфата входит в состав окислительных декарбоксилаз кетокислот. Методы определения тиамина основаны на его спектральных свойствах. Максимум поглощения тиамина составляет λ = 250 нм. Продукт окисления тиамина красной кровяной солью в щелочной среде – тиохром флюоресцирует при ультрафиолетовом освещении.
Рибофлавин, или витамин B2, идентифицирован в природных объектах в виде четырех форм, одна из которых свободный рибофлавин, остальные связаны с нуклеотидами (флавинмононуклеотид, флавинадениндинуклеотид) и с белком. Рибофлавин входит в состав простетической группы флавиновых ферментов, осуществляющих реакции дегидрирования. Окисленная форма рибофлавина так же, как и тиамина, способна флюоресцировать в ультрафиолетовом свете. Максимум поглощения рибофлавина лежит в области λ = 225 нм.
Цель работы – качественное и количественное определение витаминов B1 и B2 в вытяжках из вегетативных частей растений, корнеплодов и плодов.
Реактивы и материалы: 0,1 н. раствор H2SO4, 10 % трихлоруксусная кислота (ТХУ), 15 % раствор NaOH, 1 % раствор красной кровяной соли K3[Fe(CN)6], бромид тиамина (хлорид тиамина), рибофлавин, Na2S2O4 • H2O либо NaBH4, 0,01 н. раствор NaOH, металлический цинк, пепсин либо трипсин, изоамиловый спирт, изобутиловый спирт, этиловый спирт, диэтиловый эфир, дистиллированная вода, фарфоровые ступки с пестиками, конические колбы объемом 100 мл, водяная баня, мерные цилиндры, пробирки, делительные воронки объемом 50 мл, бумажные фильтры, термостат, центрифуга, флюоресцентный спектрофотометр.
Ход работы
1. Извлечение витаминов
1. К навеске измельченного растительного материала массой 10–20 г прилить небольшое количество (2–5 мл) 0,1 н. раствора Н2SO4, после чего все тщательно растереть в ступке.
2. Растертую массу поместить в коническую колбу на 100 мл, сюда же прибавить 30 мл 0,1 н. раствора Н2SO4. Содержимое колбы следует выдержать в течение 20 минут на кипящей водяной бане для экстрагирования витаминов.
3. После завершения процедуры экстрагирования смесь охладить и добавить в колбу 30 мг пепсина либо трипсина для перевода витамина в свободное состояние.
4. Объем гомогената довести до 50 мл раствором серной кислоты. Провести ферментацию в течение 20 ч. в термостате при 38 ºС.
5. По истечении указанного времени вытяжку центрифугировать при 5000 об/мин и отфильтровать под вакуумом. К получившейся надосадочной жидкости прилить равный объем 10 % ТХУ и выпарить до 10 мл. Продолжить выпаривание вытяжки, удаляя избыток ТХУ 2-3-кратным промыванием смесью этилового спирта и диэтилового эфира (1:1), до объема 5 мл.
Далее полученную вытяжку разделить на две части: одна идет на определение тиамина, а вторая – рибофлавина.
2. Получение тиохрома
1. В делительные воронки объемом 50 мл налить 2,5 мл полученной вытяжки. В одну из воронок (контрольную) прилить 1,5 мл 15 % раствора NaOH, перемешать и добавить 6 мл изобутилового спирта.
2. В опытные воронки прибавить по 1,5 мл 0,04 % раствора красной кровяной соли в 15 % растворе NaOH, все содержимое перемешать и прилить 6 мл изобутилового спирта.
3. Делительные воронки закрыть пробками и сильно встряхивать в течение 1 мин. Дать смеси отстояться, затем нижний слой следует слить. Для просветления спиртового слоя прилить 1 мл этилового спирта, встряхнуть, дать отстояться и полученный прозрачный раствор слить в пробирку для последующего спектрофотометрического анализа.
3. Флюоресцентная спектроскопия растворов тиохрома и рибофлавина
1. Полученные опытные растворы тиохрома перелить в кюветы и провести измерение интенсивности флюоресценции (длина волны возбуждения λ = 250 нм). Максимум флюоресценции для тиохрома составляет λ = 495-500 нм.
Аналогично провести процедуру измерения интенсивности флюоресценции для растворов рибофлавина (длина волны возбуждения равна λ = 225 нм). Регистрировать максимум флюоресценции рибофлавина в области λ = 525-530 нм.
Учитывая, что спектральные свойства окисленных и восстановленных форм тиамина и рибофлавина изменяются, провести их сравнительный флюоресцентный анализ. Рибофлавин окисляется 4 % раствором перманганата калия, который добавляется к вытяжке по каплям до исчезновения красноватой окраски, либо концентрированной HCl в присутствии металлического цинка. Восстановление окисленных форм тиамина и рибофлавина осуществить либо гидросульфитом натрия (Na2S2O4 • H2O) либо боргидридом натрия (NaBH4), добавляя эти восстановители в количестве 0,1–0,2 г на 1–2 мл раствора витаминов.
Результаты оформить в виде таблицы и сделать выводы.
| Витамин | Возбуждение флюоресценции, нм | Интенсивность флюоресценции, отн. ед. | ||
| окисленная форма | восстановленная форма | окисленная форма | восстановленная форма | |
| В1 | ||||
| В2 | ||||
4. Количественное определение витаминов В1 и В2
Для количественной оценки содержания витаминов в растительных образцах следует получить калибровочные графики зависимости интенсивности флюоресценции от концентрации. С этой целью необходимо:
1. Приготовить стандартные растворы витаминов тиамина и рибофлавина (10 мг тиамина бромида (тиамина хлорида) растворить в 100 мл 0,01 н. раствора HCl, 10 мг рибофлавина растворить в 100 мл 0,01 н. раствора NaOH).
2. Провести окисление стандартного раствора тиамина. Для этого в делительную воронку налить 1 мл рабочего раствора и прибавить 4 мл воды.
(Рабочий раствор приготовить непосредственно перед окислением. Для этого взять 1 мл стандартного раствора и довести его до 100 мл дистиллированной водой 1 мл этого рабочего раствора будет содержать 1 мкг тиамина бромида).
3. В воронку прилить щелочной раствор красной кровяной соли и провести процедуру разделения и просветления растворов, как это описано выше. Окисленный и просветленный раствор перелить в пробирки для флюориметрии.
4. Приготовить рабочий раствор рибофлавина. Для этого следует взять 1 мл стандартного раствора рибофлавина и довести его до 100 мл дистиллированной водой (1 мл этого раствора будет содержать 1 мкг рибофлавина).
5. Построить калибровочные кривые для растворов тиохрома и рибофла-вина, взяв следующие концентрации для тиамина – 0,05; 0,1; 0,15; 0,2 мкг, а для рибофлавина – 0,005; 0,010; 0,020; 0,05 мкг.
По калибровочным кривым рассчитать содержание тиамина и рибофлавина (мкг/мл) в растительных образцах. Количество витаминов в исследуемом материале (мгк/г сырой массы) вычислить по формуле:
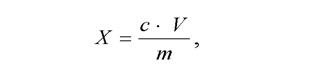
где с – содержание витамина в 1 мл вытяжки, найденное по калибро-вочной кривой; V – объем экстракта в мл; m – масса исследуемого материала в граммах.
Сделать выводы.
Контрольные вопросы
1. Содержание жиро- и водорастворимых витаминов в растительных продуктах.
2. Строение, свойства и функции витаминов в растениях.
Ход работы
1. Навеску в 1 г корня хрена, редьки или корнеплодов других растений растирают в ступке с кварцевым песком и затем водой количественно переносят в посуду на 10 мл и объем вытяжки доводят до метки.
2. Вытяжку фильтруют через складчатый фильтр.
3. В колбу на 25 мл наливают 2 мл 1 %-ного раствора бензидина, 2 мл 3%-ного раствора перекиси водорода и 1 мл приготовленного ферментного препарата; содержимое колбочки перемешивают и оставляют стоять 3 минуты (по песочным часам).
4. Через 3 минуты в колбу прибавляют 10 мл 30%-ного раствора NaOH и перемешивают.
Выпавшее в осадок окрашенное соединение растворяют прибавлением 10 мл абсолютного спирта, а затем полученный раствор колориметрируют по стандартному раствору.
Стандартный раствор готовят в мерной колбе на 25 мл. В колбу наливают 2 мл 1 %-ного раствора бензидина и 3 мл 0,01 н. раствора КМnO4.
Через 10 минут, когда появившаяся сначала зеленая окраска превратится в темно-красную, прибавляют 10 мл 30%-ного раствора NaOH и 10 мл абсолютного спирта.
Вычисление результатов. За единицу пероксидазы принимают такую ее активность, при которой препарат пероксидазы за 3 минуты дает окрашивание, равное стандарту.
Активность пероксидазы (С) в принятых единицах на 1 г растительной ткани будет равна:
С = h 1 . 10
h2
где h1 – толщина слоя образцового раствора; h2 – толщина слоя испытуемого раствора.
Оборудование и реактивы: 1) колориметр, 2) колбы на 10 и 25 мл, 3) пипетки, 4) песочные часы на 3 мин., 5) фарфоровая ступка, 6) спирт абсолютный, 7) NaOH 30%-ный, 8) Н2О2 3%-ная, 9) раствор бензидина в 1%-ной ледяной уксусной кислоте, 10) 0,01 н. КМnО4
Ход работы
1. Навеску 5 г свежего растительного материала растирают в ступке со стеклянным песком и 0,3 г СаСО3
2. Добавляют 20 мл воды и снова тщательно растирают до получения однородной массы.
3. После этого растертую массу водой количественно переносят в мерную колбу на 100 мл и доводят объем болтушки до метки.
4. Через 30–40 минут смесь фильтруют или центрифугируют.
5. Две порции чистого фильтрата или центрифугата (по 20 мл) помещают в колбы на 100 мл. Одну из колб кипятят 2–3 минуты для инактивации фермента и затем охлаждают.
6. В обе колбы приливают по 20 мл воды и по 3 мл 1%-ного раствора перекиси водорода. Время инкубации от 20 до 30 минут.
7. По окончании инкубации к содержимому обеих колб добавляют 4–5 мл 10%-ного раствора H2SО4 и титруют 0,1 н. раствором КМnO4 до слабо-розовой окраски, не исчезающей в течение минуты.
По разности между контрольным и опытным титрованием определяют количество разложившейся перекиси водорода за время инкубации в расчете на 1 г исходного растительного вещества.
x = (а – б) · Т · 1,7
Н
где х – активность каталазы в миллиграммах Н2О2, разложившейся за время инкубации на 1 г растительного вещества; а – миллилитров 0,1н. раствора КМnO4, израсходованного на холостое титрование; б – миллилитров 0,1 н. раствора КМnO4, пошедшего на опытное титрование; Т – поправка к титру 0,1 н. КМп04; 1,7 – количество миллиграммов Н2О2, соответствующее каждому миллилитру 0,1 н. КМnO4; Н – навеска растительного материала.
Оборудование и реактивы: 1) колбочки на 100 мл; 2) мерные колбочки на 100 мл; 3) пипетки; 4) бюретка; 5) фарфоровая ступка.
Ход работы
1. 20 г мякоти тыквы растирают в ступке с 10–15 мл 0,15 М фосфатного буфера с рН около 7.
2. Растертую массу переносят в широкогорлую мерную колбу на 50 мл с доведением объема взвеси до метки фосфатным буфером.
3. Через 1 час настаивания содержимое колбы фильтруют и фильтрат используют в качестве препарата фермента.
4. Две порции препарата по 10 мл помещают в колбочки на 50 мл и одну из них нагревают до кипения и кипятят 1–2 минуты для инактивации фермента, а затем охлаждают.
5. Колбы ставят в термостат при температуре 37°С и после добавления 10 мл раствора аскорбиновой кислоты инкубируют смесь в течение 30 минут.
6. Затем содержимое обеих колб нагревают до кипячения и после охлаждения доводят водой до метки.
7. По 10 мл прозрачной жидкости переносят в конические колбы емкостью 100 мл и добавляют по 5 мл 10%-ного раствора KJ, 10 мл 5%-ной НС1, 10 капель 1%-ного раствора крахмала и 10 мл 0,01 н. KJО3 в каждую из колб.
8. Через пять минут после прибавления последнего реактива растворы титруют 0,001 н. Na2S2О3 до исчезновения синей окраски.
Вычисление результатов. Результаты определения проводят по следующей формуле:
х = (а - б) · Т · 0,088
Н
где х – активность аскорбатоксидазы, выраженная в миллиграммах окисленной аскорбиновой кислоты под действием фермента из 1 г растительной ткани за период инкубации;
а – миллилитров 0,001 н. раствора гипосульфита, израсходованного на оттитровывание избытка йода в опытном определении;
б – миллилитров 0,001 н. раствора гипосульфита, израсходованного на титрование контрольной пробы;
Т – поправка к титру 0,001 н. Na2S203;
0,088 – количество миллиграммов аскорбиновой кислоты, соответствующее каждому миллилитру 0,001 н. Na2S203;
Н – навеска вещества, соответствующая объему препарата, взятого для титрования (в г).
Оборудование и реактивы: 1) термостат; 2) фарфоровая ступка; 3) мерные колбы на 25 и 50 мл; 4) конические колбы на 100 мл; 5) 0,01 н. KJО3; 6) KJ 10%-ный; 7) 0,001 н. Na2S2О3; 8) крахмал, 1%-ный раствор; 9) фосфатный буфер, 0,15 М с рН=7,0; 10) аскорбиновая кислота (раствор с содержанием 1 мг в 1 мл).
Определение активности тирозиназы
Тирозиназа относится к классу оксидоредуктаз, к группе фенолоксидаз. Этот фермент, в отличие от других ферментов этой группы, обладает групповой (относительной) специфичностью и катализирует окисление не только монофенолов, но и полифенолов. Тирозиназа широко распространена в растительных организмах. По своей природе она является медьсодержащим протеидом.
За каталитическим действием тирозиназы можно наблюдать по реакции окисления тирозина кислородом воздуха в присутствии препарата тирозиназы. В процессе окисления из тирозина образуется красный пигмент гелохром, а затем темный пигмент меланин.
Ход работы
1. Клубень картофеля растирают на терке и навеску 1–2 г растительной массы заливают 5 мл воды и тщательно перемешивают.
2. Полученную взвесь фильтруют через 2 слоя марли и фильтрат используют как препарат фермента.
3. В две пробирки наливают по 1 мл препарата фермента.
Содержимое одной из них (для «холостого» определения) нагревают до кипения и кипятят 1–2 минуты, а затем охлаждают.
4. В обе пробирки добавляют по 1 мл 1%-ного раствора тирозина и при периодическом (через 3 минуты) перемешивании встряхиванием реакционную смесь инкубируют при температуре 40°С.
За окраской жидкости наблюдают в пробирках. Жидкость в пробирке с активной вытяжкой из картофеля постепенно становится сначала розовой, затем бурой и, наконец, черной. В «холостой» пробе с инактивированным ферментом цвет жидкости в пробирке не изменяется.
Результаты наблюдения фиксируют в тетради и делают выводы.
Оборудование и реактивы: 1) терки; 2) пробирки; 3) пипетки; 4) термостат водяная баня; 5) тирозин, 1%-ный раствор.
Определение гидролитической активности липазы
Липазы, относящиеся к группе эстераз, являются ферментами, катализирующими расщепление сложноэфирной связи между глицерином и жирными кислотами в жире. В результате действия липаз жир расщепляется на глицерин и свободные жирные кислоты.
Липазы распространены во многих растительных объектах, они присутствуют почти во всех органах и тканях растений, но больше всего их в семенах, особенно в семенах масличных растений. Активность липаз из разных растений различна, они отличаются также по оптимуму рН, температурному оптимуму и ряду других свойств. При прорастании семян липазы расщепляют содержащийся в них жир и продукты распада подвергаются затем дальнейшим превращениям.
Активность липаз имеет большое значение при хранении муки и круп, особенно содержащих много жира. При увеличении влажности этих продуктов и повышенной температуре хранения липазы быстро расщепляют жиры с образованием свободных жирных кислот, что приводит к повышению кислотности продуктов и их быстрой порче, прогорканию.
Принцип метода. В качестве источника фермента используется тонкоразмолотый испытуемый материал (чаще всего семена масличных растений) или этот же материал, но предварительно обезжиренный эфиром или ацетоном. Так как под действием липаз из жира освобождаются жирные кислоты, активность липаз определяют по увеличению кислотности в реакционной смеси при прямом титровании щелочью RCOOH + NaOH → RCOONa + Н2О, и показателем активности служит количество щелочи, требующееся для нейтрализации образовавшихся жирных кислот. Идеальным субстратом при изучении действия липазы из какого-либо объекта должен был бы являться жир, полученный из того же объекта. Однако в качестве субстратов действия липаз можно пользоваться и другими естественными жирами и маслами различного происхождения.
Ход работы
1. Две навески (по 3 г) размолотых семян тщательно растирают в ступке с песком и переносят в конические колбы емкостью 100 мл с притертыми пробками.
2. Ступку смывают 2–3 раза небольшим количеством (по 2–3 мл) воды и в колбы добавляют по 1 мл чистого подсолнечного масла.
В связи с тем, что оптимум рН большинства растительных липаз равен 4,5–5,0, в реакционную колбу добавляют еще 5 мл ацетатного буфера с рН=4,7 (ацетатный буфер с рН=4,7 готовят смешиванием одного объема 1 н. СН3СООН и одного объема 1 н. CH3COONa) и несколько капель толуола.
3. Смесь в колбах тщательно перемешивают на механической мешалке, после чего ставят на 20–24 часа в термостат при температуре 30°С или оставляют
при комнатной температуре. За этот срок значительная часть введенного подсолнечного масла расщепляется липазами, имеющимися в семенах.
Одновременно с используемыми пробами ставят контрольные.
4. Для контрольных проб берут также по две навески растертого материала и поступают с ними таким же образом, но перед тем, как ставить в термостат, их предварительно кипятят в течение 3–5 минут для инактивации ферментов.
5. По истечении 20–24 часов в конические колбы добавляют по 25 мл спирта и 15–25 мл эфира.
6. Содержимое колб встряхивают, дают отстояться и титруют 0,1 н. NaOH в присутствии 0,5 мл 1%-ного спиртового раствора тимолфталеина (для неокрашенных экстрактов в качестве индикатора можно применять фенолфталеин).
Вычисление результатов. Активность липаз выражают в миллилитрах 0,1 н. NaOH на 10 г семян. Расчеты можно проводить по следующей формуле:
х = (а · Т – б · Т) · 10
Н
где х – активность липазы;
а – миллилитров 0,1 н. NaOH, израсходованных для титрования опытного образца; Т – поправка к титру 0,1 н. NaOH; б – миллилитров 0,1 н. NaOH, израсходованных для титрования контрольного (предварительно прокипяченного) образца; Н – навеска семян.
Приготовление чистого масла. 300 г подсолнечного масла взбалтывают в делительной воронке с 2%-ным NaOH и дают отстояться. Щелочной раствор сливают. Промывают в воронке несколько раз дистиллированной водой (до отрицательной реакции с фенолфталеином). Затем отстаивают и сушат, пропуская через колонку с хлористым кальцием.
Оборудование и реактивы: 1) ступки фарфоровые диаметром 10 см; 2) колбы конические емкостью 100 мл с притертыми пробками; 3) чистое подсолнечное масло; 4) ацетатный буфер (рН=4,7); 5) этиловый спирт; 6) эфир; 7) толуол; 8) тимолфталеин или фенолфталеин, 1%-ный спиртовый раствор. 9) 0,1 н. NaOH.
Контрольные вопросы
1. Особенности строения, свойства и функции ферментов.
2. Классификация ферментов (с примерами).
3. Механизм ферментативного катализа.
4. Зависимость активности ферментов от: температуры реакции, кислотности среды, концентрации субстрата.
5. Активаторы ферментов.
6. Ингибиторы ферментов.
7. Характеристика ферментов: а) 1 класса, входящие в состав зерна и продуктов его переработки; б) 2 класса; в) 3 класса.
8. Промышленное использование растительных ферментов.
9. Иммобилизация ферментов.
Соединений
Ход работы
В качестве контроля берут 20 мл прокипяченной воды. Воду кипятят для удаления углекислоты и затем охлаждают.
1. 20 мл испытуемого и 20 мл контрольного раствора переносят в конические колбы, куда добавляют по 3 капли метилового красного.
2. Содержимое колб титруют 0,1 н. NaOH до золотисто-желтой окраски.
3. К нейтрализованным растворам прибавляют по 10 мл формольной смеси. Эту смесь готовят так: к 50 мл 40%-ного формалина добавляют 1 мл 1%-ного спиртового раствора фенолфталеина и смесь доводят 0,2 н. щелочью до слабо-розового окрашивания.
4. После добавления формольной смеси растворы титруют 0,2 н. NaOH.
При этом следует внимательно следить за изменением окраски. Сначала розовая окраска переходит в желтую (изменение цвета от индикатора метилового красного, который дает желтое окрашивание при рН=6,2), а потом в розовую (фенолфталеин дает слабо-розовое окрашивание при рН 8,2–8,3).
5. Затем добавляют еще 1–2 капли 0,2 н. NaOH до ясно-красного окрашивания (рН=8,8) и еще 2 капли щелочи до интенсивно красной окраски (рН=9,1). Оба раствора титруют до одинакового тона.
При использовании в качестве индикатора тимолфталеина формольная смесь готовится так: к 50 мл 40%-ного формалина прибавляют 25 мл спирта, 5 мл спиртового раствора тимолфталеина (50 мг в 100 мл 96%-ного спирта) и 0,2 н. NaOH до слабо-зеленой или синеватой окраски.
Дальнейший ход анализа должен быть таким же, как и при использовании формольной смеси с фенолфталеином, но контрольная и опытная пробы должны быть оттитрованы 0,2 н. NaOH до ярко-синего окрашивания (рН=9,7).
Вычисление результатов. 1 м-экв. аминного азота соответствует
1м-экв. щелочи. При использовании 0,2 н. NaOH 1 мл щелочи соответствует 14 . 0,2 = 2,8 мг N. Расчет ведут по формуле:
х = (а - b ) . T . 2,8
где х – количество аминного азота, содержащееся в 20 мл исследуемого раствора (мг); а – количество миллилитров 0,2 н. NaOH, израсходованного на титрование испытуемого образца; б – количество миллилитров 0,2 н. NaOH, израсходованного на титрование контроля; Т – поправка к титру 0,2 н. NaOH.
Оборудование и реактивы: 1) колбы конические емкостью 100 мл; 2) пипетки; 3) 0,1 н. NaOH; 4) 0,2 н. NaOH; 5) метиловый красный; 6) фенолфталеин; 7) тимолфталеин; 8) формалин 40%-ный; 9) спирт 96%-ный.
Проведение анализа.
Подготовка проб для анализа.
Пробы (за исключением растений семейства крестоцветных), измельчают с помощью измельчителя тканей РТ-1 (РТ-2) (возможно применение для этих целей мезгообразователя МЛ-1, терки, механической или электрической мясорубки, пищевого измельчителя). Зеленые культуры режут ножницами или ножом до частиц размером 0,5 - 1,0 см или измельчают на мясорубке. 10.0 г измельченного материала взвешивают с точностью до второго десятичного знака, помещают в стакан гомогенизатора или измельчителя тканей РТ-1, добавляют 50 мл экстрагирующего раствора.
10,0 г взвешенного до первого десятичного знака измельченного на терке, мясорубке или мезгообразователе материала помешают в стакан вместимостью около 100 мл, добавляют 50 мл экстрагирующего раствора и перемешивают с помощью мешалки в течение 3 минут.
При анализе материала, содержащего твердые ткани, и отсутствии гомогенизатора пробу массой 10,0 г растирают в ступке с прокаленным песком или битым стеклом до однородной массы, помещают в стакан вместимостью около 100 мл, добавляют 50 мл экстрагирующего раствора и перемешивают с помощью мешалки в течение 3-х минут.
При анализе растений семейства крестоцветных (капуста, редис) 10,0 г измельченного материала взвешивают с точностью до первого десятичного знака, помешают в стакан вместимостью 100 мл, наливают 50 мл экстрагирующего раствора, перемешивают с помощью мешалки в течение трех минут. Затем при перемешивании добавляют по каплям (2-3 капли) 33%-ный раствор перекиси водорода до обесцвечивания раствора.
В суспензии, приготовленной одним из описанных выше способов, измеряют концентрацию нитрат-ионов.
Для получения сока пробы, материал пропускают через электрическую или механическую соковыжималку. Полученный сок собирают в одну емкость и перемешивают.
При анализе всех культур, кроме растений семейства крестоцветных (капуста, редис), от полученного сока с помощью пипетки отбирают аликвотную часть объемом 10,0 мл, измеренным с точность до 0,1 мл, помещают
ее в стакан вместимостью 100 мл, добавляют 50 мл экстрагирующего раствора, перемешивают и в полученном растворе измеряют концентрацию нитрат-ионов.
При анализе растений семейства крестоцветных (капуста, редис) в стакан вместимостью 100 мл помещают 10,0 мл сока, измеренного с точность до 0,1 мл, добавляют 50 мл экстрагирующего раствора, перемешивают в течение 3-х минут. Затем при перемешивании добавляют по каплям (2-3 капли) 33%-ный раствор перекиси водорода до обесцвечивания раствора, после чего измеряют концентрацию нитрат-ионов.
Отбор и подготовка проб для анализа.
Общие требования к отбору проб. Пробы растительной продукции должны быть представительными и достоверно характеризовать качественный состав сельскохозяйственной продукции, включая содержание в них нитратов на нолях в предуборочный период и в партиях растительной продукции при ее реализации по назначению. В соответствии с этими задачами пробу следует отбирать либо в поле на заключительном этапе созревания культур («на корню»), либо из партий собранной растительной продукции, готовой к реализации.
Отбор растительных проб «на корню» проводят в утренние часы после схода росы до наступления жары (с 7 до 11 часов). Пробы нельзя отбирать во время дождя и полива или сразу же после них.
Для составления средней пробы используют метод прохода по диагонали с отбором точечных проб через равные расстояния. Для формирования смешанной пробы нельзя брать растения, имеющие площадь питания большую или меньшую по сравнению с остальными растениями (из крайних борозд, гряд, рядов, из гнезд с выпавшими растениями, а также из соседних с ними гнезд), отставшие в развитии или слишком мощные отдельные растения. При этом плоды должны быть без признаков заболевания, механических повреждений, без налипшей почвы.
Перед отбором проб растений для определения нитратов следует подготовить: чистые мешки или сетки для упаковки и перевозки отобранных проб корнеклубнеплодов, бахчевой продукции; ящики для овощной продукции; полиэтиленовые или целлофановые пакеты с упаковочными резинками для отбора смешанных проб, чтобы не допустить потери влаги.
Отобранные пробы сопровождаются этикеткой и актом отбора проб сельскохозяйственной продукции в 2-х экз.
Отбор проб с поля («на корню»)
Картофель. С участка площадью до 10 га по диагонали выкапывают без выбора не менее 20 кустов, расположенных через равные промежутки.
Клубни отряхивают от земли и отделяют от столонов. Из каждого кусга отбирают ПО одному среднему и крупному клубню».
Капуста белокочанная, краснокочанная и цветная. Отбирают не менее 10 типичных кочанов, равномерно расположенных по диагонали с каждого участка площадью 5 га. С кочанов снимают верхние кроющие листья.
Овощные корнеплоды. С участка площадью до 3 га по диагонали через равные промежутки отбирают не менее 20 корнеплодов столовой свеклы, моркови, петрушки, сельдерея, редьки, редиса.
Томаты, огурцы, баклажаны, перец сладкий. Томаты в зонах товарного производства являются многосборовой культурой и пробы плодов для анализа стимают по достижении ими съемной зрелости. В пробу должно войти не менее 20 типичных плодов отобранных по диагонали с площади 3 га.
Методы отбора проб огурцов, баклажанов, перца аналогичны отбору проб томатов.
Лук репчатый. Отбор проб лука репчатого проводят, когда большая часть мелких луковиц уже созрела и имеет усыхающие листья, а крупные луковицы – частично неполегшие зеленые листья. Выкапывают не менее 20 луковиц с участка 5 га равномерно по диагонали.
Кабачки, тыква, патиссоны. Отбор проб проводят по мере созревания плодов в период массового сбора. В среднюю пробу должно войти не менее 10 типичных плодов с растений, расположенных равномерно по диагонали поля с участка до 5 га.
Листовые (зеленые овощи – лук-перо, салат, шпинат, кориандр, петрушка, сельдерей, щавель, укроп и др.). Отбор проб проводят по диагонали участка до 1 га на 20 точек массой не менее 0,5 кг через равные промежутки.
Овощные культуры защищенного грунта. Общее количество плодов в объединенной пробе должно быть для огурца короткоплодного – 35– 40, для огурца длинноплодного – 20– 25, для томата – 40– 50, для перца – 60– 80.
Общая масса объединенной пробы плодов должна составлять не менее 3 кг, для длинноплодного огурца не менее 10 кг. Масса объединенной пробы листовых овощей должка быть не менее 1,5 кг.
В ангарных теплицах площадью 1000 м2 при отборе проб теплицу визуально делят двумя диагоналями и отбор продукции проводят с растений, равномерно расположенных по диагонали, за исключением подлежащих выбраковке (с при пиками сильных повреждений).
При количестве плодов в объединенной пробе – 40 штук по каждой диагонали отбирают с 10 растений по 2 плода (по 1 с каждого из двух верхних ярусов) и по 20 растений листовых овощей.
В блочной теплице площадью 10000 м2 выбирают три полусекции (пробных площадок) площадью около 1000 м2 в начале, середине и конце теплицы.
Общее число плодов (растений), которые необходимо отобрать с 10000 м2 делят на 3 пробные площадки, с которых отбор проб производят, как для ангарных теплиц.
Для многосборовых культур (томаты, огурцы, перец) пробы отбираются из сформированных партий собранной продукции.
Семечковые культуры (яблоки, груши). Представительную пробу семечковых культур отбирают с участка площадью не более 2 га. С каждого четвертого дерева, проходя по диагонали участка, снимают плодосъемником
плода с южной и северной стороны дерева.
Подготовка проб для анализа.
Пробы к анализу готовят следующим образом:
Картофель. Клубни моют водой, вытирают чистой тканью досуха и разрезают крестообразно вдоль оси «столон – ростовая часть» на 4 равные части. От каждого клубня берут четвертую часть, отобранный материал используют для анализа.
Свекла и другие корнеплоды. Корнеплоды моют водой, вытирают чистой тканью досуха, срезают шейку и тонкий конец корня и разрезают крестообразно вдоль вертикальной оси на 4 равные части. Доли, представляющие четвертую часть от каждого корнеплода используют для анализа.
Капуста. Кочаны разрезают крестообразно вдоль вертикальной оси на 4 части или 8 равных частей и берут соответственно по 1/4 или 1/8 части от каждого кочана в пробу для анализа. При этом отбрасывают верхние несъедобные листья и остаток кочерыжки.
Луковичные растения. Отбрасывают несъедобные части. С луковиц удаляют чешуи, срезают и отбрасывают основания корня и сухую шейку, разрезают их крестообразно вдоль. Кабачки – плоды моют водой, вытирают чистой нитью досуха, удаляют плодоножки и разрезают крестообразно вдоль оси на 4 равные части. От каждого плода в пробу для анализа берут по 4 части.
Томаты, огурцы. Плоды разрезают вдоль оси на сегменты шириной 6– 8 см по окружности плода и в пробу для анализа от каждого плода берут по 2– 4 сегмента с противоположных сторон таким образом, чтобы в их число попали затемненные и освещенные солнцем части отобранных частей плода снимают верхний слой, не употребляемый в пищу, удаляют семена.
Бахчевые культуры. Плоды разрезают вдоль оси па сегменты шириной 6 – 8 см по окружности плода и в пробу для анализа от каждого плода берут по 2–4 сегмента с противоположных сторон таким образом, чтобы в их число попали затемненные и освещенные солнцем части. С отобранных частей плода снимают верхний слой, не употребляемый в пищу, удаляют семена.
Перец сладкий. Плоды моют водой, вытирают чистой тканью досуха, разрезают крестообразно вдоль оси на 4 равные части и берут в пробу для анализа по 1/4 части от каждого плода. При этом вырезают и отбрасывают семена и остаток плодоножки.
Зеленые овощи (салат, шпинат, капуста салатная, петрушка, щавель, сельдерей, кинза, укроп и т.д.). Обрезают и отбрасывают несъедобные части растений. Растения моют водой и подсушивают сначала между листьями фильтровальной бумаги, или слоями чистой ткани, а затем на воздухе.
Яблоки, груши. Плоды моют водой, вытирают чистой тканью досуха, разрезают крестообразно вдоль оси на 4 равные части и берут в пробу для анализа по 4 части от каждого плода. При этом вырезают и отбрасывают остаток семенного гнезда и плодоножку.
Виноград. Ягоды винограда отделяют от веток, моют водой и сушат на
листе фильтровальной бумаги.
Измерение концентрации нитрат-ионов.
Подготовка к работе прибора «Эксперт-001» и электродов.
Включите блок питания прибора в сеть.
Нажмите клавишу «вкл.», расположенную на панели прибора, и удерживайте ее в нажатом положении в течение 2–4 секунд.
На дисплее индикатора появляются последовательные надписи:
Эконикс-Эксперт-001-3-0, затем АКК =…%, выбор режима рН-метр-иономер. Нитратный электрод подключите к разъему «изм», а электрод сравнения к разъему «всп», установите электроды в штатив.
Выбор режима измерений (продукта для анализа). Нажать кнопку Ф1 и кнопками выбрать наименование группы продуктов, в которой будет производиться измерения нитратов: картофель – капуста – сок картофеля – сок свеклы – сок капусты – сок огурцов. Далее нажать кнопку «ввод».
На дисплее снова появится надпись: Выбор режима рН-метр-иономер.
Выберите соответствующий режим определения нитратов и нажмите кнопку «изм». Начнется измерение и отображение измеренной концентрации нитрат-иона в анализируемой пробе в мг/кг, например:
Показания прибора считывают после того, как отображаемое значение концентрации перестанет изменяться. Как правило, для этого достаточно 30 сек. На этом процесс измерений в этой пробе закончен.
Для измерения содержания нитратов в следующей пробе достаточно вынуть электроды из стаканчика, промыть их дистиллированной водой, осушить фильтровальной бумагой и опустить в стаканчик со следующей подготовленной для анализа пробой.
Полученные результаты сравнить с данными таблицы и сделать выводы о количестве нитратов в исследуемых растительных образцах.
Допустимые уровни содержания нитратов в продуктах растительного происхождения (СанПиН 42-123-4619-88 от 30 мая 1988 г.)
| Пищевой продукт | Содержание нитратов, мг/кг | |
| из открытого грунта | из защищенного грунта | |
| Картофель | 250 | - |
| Капуста белокочанная ранняя (до 1 сентября) поздняя | 900 500 | - |
| Морковь ранняя (до 1 сентября) поздняя | 400 250 | - - |
| Томаты | 150 | 300 |
| Огурцы | 150 | 400 |
| Свекла столовая | 1400 | - |
| Лук репчатый | 80 | - |
| Лук-перо | 600 | 800 |
| Зеленые культуры (салат, шпинат, щавель, капуста салатная, петрушка, сельдерей, кинза, укроп и др.) | 3000 | 3000 |
| Дыни | 90 | - |
| Арбузы | 60 | - |
| Перец сладкий | 200 | 400 |
| Кабачки | 400 | 400 |
| Тыква (для изготовления консервов для питания детей) | 200 | - |
| Виноград столовых сортов | 60 | - |
| Яблоки | 60 | - |
| Груши | 60 | - |
Контрольные вопросы
1. Содержание нитратов в растениях. 2. Этапы восстановления нитратов в растениях. Факторы, влияющие на восстановление нитратов.
3. Причины накопления избыточного количества нитратов в растениеводческой продукции.
4. Характерные особенности отравления продукцией с высоким содержанием нитратов.
5. Возможные пути снижения избыточного количества нитратов в сельскохозяйственной продукции.
Ход работы
Взвесить 10 –20 г свежих размельченных плодов. Перенести навеску в фарфоровую ступку и тщательно растереть с 1-2 г кварцевого песка до однородной массы. Растертую массу количественно перенести в коническую колбу на 250 мл, залить 100 мл горячей дистиллированной воды (80оС) и нагревать на водяной бане в течение 1 ч при 80оС. Затем содержимое колбы охладить и отфильтровать через воронку Шотта. Довести объем экстракта до 100 мл. Пипеткой взять 20 мл вытяжки и перенести в чистую коническую колбу, туда же добавить 2-3 капли фенолфталеина до розового окрашивания. Оттитровать вытяжку 0,1 н. раствором щелочи. Кислотность исследуемого объекта (Х, %) вычислить по формуле:
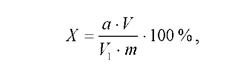
где а - количество 0,1 н. щелочи, пошедшей на титрование, в мл; V - общий объем вытяжки; V 1 - объем вытяжки, взятой для титрования; т- масса навески в граммах.
Если результат хотят выразить для какой-либо из главных органических кислот, то Х умножают на определенный расчетный коэффициент. Согласно А. И. Ермакову и др., 1 мл 0,1 н раствора щелочи пошедшей на титрование, соответствует 7,5 мг винной, 6,7 мг яблочной, 6,4 мг лимонной, 4,5 мг щавелевой кислот.
Результаты оформить в виде таблицы и сделать выводы.
| Объект | Количество щелочи, пошедшей на титрование, мл | Кислотность, % |
Определение общей кислотности
В плодах и овощах, а также в листьях многих растений часто накапливается значительное количество свободных кислот. Определение общего содержания кислот имеет большое значение при использовании плодов и овощей в пищу, при их консервировании, а также при изучении накопления и распада кислот в растениях. Величину общей кислотности можно определить алкалиметрическим, а также ацидиметрическим титрованием с использованием соответствующих индикаторов или потен-циометрически.
Принцип метода. Кислоты извлекаются из измельченного растительного материала в результате нагревания с содой при температуре 80-90° в течение 30 минут. Извлеченные кислоты оттитровывают раствором щелочи. Общее количество кислот обычно пересчитывают на яблочную кислоту.
Ход работы
1. Свежие плоды, овощи или листья растений тщательно измельчают на терке, в мясорубке или в ступке. Растительную массу перемешивают.
2. Берут навеску 20 г и переносят ее без потерь в широкогорлую колбу объемом 200 мл.
3. В колбу приливают около 150 мл дистиллированной воды и выдерживают в течение 30 минут в водяной бане при температуре 80-90°.
4. Затем колбу охлаждают водопроводной водой, доводят до метки и фильтруют в сухой стакан или колбу.
Полученный фильтрат служит для определения общей кислотности.
5. 50 мл фильтрата, содержащего кислоты, переносят в коническую колбу емкостью 200-250 мл и добавляют в нее 50 мл 1 н. NaOH.
6. Колбу нагревают на водяной бане при 80-90° в течение 10 минут.
При нагревании раствора с избытком щелочи ангидриды кислот расщепляются.
7. Затем в колбу добавляют 50 мл 1,2 н. НС1 и снова нагревают на бане в течение 10 минут.
При нагревании кислотного раствора из него удаляется углекислота. Затем раствор в колбе охлаждают.
Наряду с определением свободной кислотности в анализируемом образце проводят «холостое» определение. Для этого вместо раствора берут 50 мл дистиллированной воды, добавляют 50 мл 1,0 н. NaOH, нагревают на бане, затем вносят 1,2 н. НС1 и снова нагревают.
8. После охлаждения растворов приступают к титрованию. Для этого в обе колбы добавляют по нескольку капель спиртового раствора фенолфталеина и титруют 0,1 н. NaOH до ярко-розового оттенка раствора.
Если раствор сильно окрашен и переход окраски раствора при добавлении щелочи определить трудно, то при титровании можно использовать лакмусовую бумажку. Капли жидкости из колбы при титровании переносят на кусочек фильтровальной бумаги и наблюдают изменение окраски.
Вычисление результатов. При расчете необходимо учитывать количество миллилитров добавленных щелочи и кислоты. Разницу в титре 1 н. NaOH и 1,2 н. НС1 следует принимать во внимание при проведении «холос- того» определения (х) в таком же объеме жидкости.
Количество миллиэквивалентов кислот, содержащихся в 100 мл раствора (в 10 г образца), равняется:
100 X у – х
50 10
где у - число миллилитров 0,1 н. NaOH, затраченное на титрование исследуемого образца;
х - число миллилитров 0,1 н. NaOH, затраченное на титрование контроля.
Для вычисления процентного содержания кислот необходимо полученный результат умножить на 10, а для перечисления на яблочную кислоту умножить на коэффициент 0,0067 (1 м-экв. яблочной кислоты=134 : 2 = 67 мг).
Оборудование и реактивы: 1) водяные бани; 2) колбы мерные емкостью 200 мл; 3) колбы конические емкостью 200-300 мл; 4) воронки; 5) пипетки; 6) фильтры; 7) 1,0 н. и 0,1 н. NaOH; 8) 1,2 н. НС1; 9) фенолфталеин; 10) лакмусовая бумага.
Контрольные вопросы
1. Содержание в растениях органических кислот алифатического ряда.
2. Функции органических кислот в растении.
3. Характерные особенности основных органических кислот растений.
4. Обмен органических кислот у высших растений.
Ход работы
1. Навеску муки или размолотого зерна 10 г помещают в коническую колбу
на 300 см, приливают точно 100 см дистиллированной воды.
2. Тщательно размешав содержимое, колбу оставляют для возможно полного экстрагирования водорастворимых веществ на 1 час при комнатной температуре, периодически взбалтывая.
3. Затем фильтруют жидкость в сухую колбу, с возвратом первых (мутных) порций фильтрата на фильтр.
4. Берут 25 см фильтрата пипеткой и переносят в коническую колбочку на 100-150 см, прибавляют 3 капли фенолфталеина и титруют децинормальным раствором щелочи до бледно-розовой окраски.
5. Кислотность по водной вытяжке вычисляется по формуле:
a • k • C • 1000
X = P • b • (100 - W)
где k - коэффициент поправки к титру щелочи; а - объем 0,1 моль/дм раствора щелочи, пошедшего на титрование, см; С - количество воды, взятое на обработку муки, см; Р - навеска, г; W - влажность муки, %; b - количество фильтрата, взятое на титрование, см3.
Определение кислотности по водно-спиртовой вытяжке
По этому методу титруются щелочью все органические кислоты, в том числе жирные, спирторастворимые белки (проламины), аминокислоты, пептиды.
Ход работы
1. Навеску муки для размолотого зерна 2,5 г высыпают в колбу, приливают 25 см раствора спирта ю (C2H5OH) = 67 %.
2. Содержимое колбы энергично взбалтывают в течение 5 минут и фильтруют.
3. Пипеткой отбирают 25 см фильтрата, приливают к ним 3 капли фенолфталеина и титруют децинормальным раствором щелочи до розовой окраски.
4. Кислотность выражается по формуле:
a • к • C • 1000
Х = P • b • (100 - W)
где Х - кислотность в градусах на сухой вес вещества; а - количество 0,1 моль/дм раствора щелочи, пошедшего на титрование, см; k - коэффициент поправки к титру щелочи; b - количество фильтрата, взятого для титрования, см3; С - количество спирта, взятого на обработку продукта, см; Р - навеска , г; W - влажность продукта, %.
Хлебопекарные качества муки. Биохимические процессы,
протекающие при замесе теста и выпечке хлеба
Из зерна пшеницы получают муку пшеничную, хлебопекарную и макаронную. Свойства муки, обуславливающие качество хлеба, представляют собой хлебопекарные качества муки.
Хлебопекарные качества муки в основном определяются совокупностью следующих ее свойств: сахарообразующей, газообразующей, газоудерживающей и водопоглотительной способностями, цветом муки, крупностью частиц помола. Газообразующая способность, в свою очередь, зависит от количества в муке ее «собственных» сахаров, перешедших в муку из зерна и содержащихся в ней еще до замеса теста; от сахарообразующей способности муки, то есть способности образовывать в тесте мальтозу в результате действия амилаз на крахмал муки, от качества (активности) дрожжей.
В пшеничной муке содержится от 1 до 2,5 % сахара, главным образом, сахарозы, которая очень легко расщепляется под влиянием выделяемой дрожжами сахаразы (α-фруктофуранозидазы). Получающаяся смесь глюкозы и фруктозы легко сбраживается дрожжами.
Однако, этого количества сахара недостаточно, чтобы процесс брожения теста шел до конца. Собственный сахар муки используется только на самых первых этапах брожения теста, а потом его уже не хватает. На следующих этапах на первый план выступает мальтоза, образующаяся благодаря действию β-амилазы. В тесте под влиянием амилазы крахмал расщепляется с образованием мальтозы. В свою очередь, мальтоза расщепляется, образуя две молекулы глюкозы, которая сбраживается дрожжами. Если мука имеет низкую амилолитическую способность (активность), в тесте не будет образовываться достаточное количество мальтозы и глюкозы, брожение не будет протекать интенсивно и хлеб получится плохого качества, с недостаточно пористым, недостаточно разрыхленным, плотным мякишем. Мука с низкой активностью α-амилазы дает тесто, в котором образуется мало декстринов, поэтому хлеб получается с бледной коркой. Такую муку практики называют «крепкой на жар».
Газоудерживающая способность муки зависит, прежде всего, от свойств содержащихся в тесте белков, от количества и качества клейковины. В пшеничном тесте она образует тот растяжимый эластичный каркас, в котором накапливаются пузырьки углекислого газа, поднимающие тесто. Этот белковый каркас во время брожения теста постепенно расширяется.
При образовании пшеничного теста происходит, прежде всего, осмотическое связывание воды вначале свободным промежуточным белком, затем белком, окружающим отдельно лежащие крахмальные зерна и, наконец, белком, содержащемся в более крупных частицах муки, представляющих собой неразрушенные клетки эндосперма. При поглощении воды белок сильно увеличивается в объеме и постепенно образуется непрерывная структура теста, представляющая собой сетку клейковины, в которую включены крахмальные зерна и другие растворимые частицы муки.
В это же время происходит набухание крахмальных зерен, причем, чем больше содержится поврежденных крахмальных зерен, тем больше его водопоглотительная способность. Добавление соли во время замеса теста несколько снижает гидратационную способность клейковины. Вода и бродильные микроорганизмы вызывают в муке комплекс сложных биохимических превращений. Эти превращения, завершающиеся в процессе выпечки, оказывают глубокое воздействие, как на физические свойства теста, так и на вкус и аромат готового продукта.
Процесс брожения представляет собой сложный комплекс биохимических превращений, конечными продуктами которых являются углекислый газ и этиловый спирт. Во время брожения наблюдается сильное повышение активной кислотности среды за счет увеличения суммарного содержания органических кислот, большую часть которых составляют молочная и уксусная кислоты. Кроме того, образуются летучие органические кислоты, а также некоторые альдегиды и кетоны, участвующие в формировании вкуса и аромата хлеба.
Заключительным этапом приготовления хлеба является его выпечка. Под воздействием высокой температуры происходит изменение коллоидного состояния основных полимерных компонентов муки, осуществляются биохимические процессы взаимодействия различных веществ и процессы чисто физического характера. Технологическое назначение выпечки заключается в закреплении пористой структуры хлеба, достигнутой в процессе сбраживания, и в формировании вкуса и аромата хлеба, а также цвета корки.
Наиболее интенсивно биохимические процессы протекают в интервале температур 43–60°С. Происходят глубокие изменения в структуре крахмальных зерен: в результате совместного действия α- и β-амилаз на клейстеризующийся крахмал или только β-амилазы (тесто из нормальной пшеничной муки) повышается содержание водорастворимой фракции в мякише выпеченного хлеба, увеличивается также содержание растворимого в воде белка вследствие повышения атакуемости протеолитическими ферментами.
При дальнейшем повышении температуры происходит свертывание белковых веществ клейковины и клейстеризация крахмала, превращающегося в прочный студень, закрепляется пористая структура теста и формируется мякиш хлеба. В процессе выпечки заметно снижается титруемая кислотность продукта. На поверхности выпекаемых тестовых заготовок протекают биохимические процессы, существенно влияющие на качество продукта. Установлено, что под воздействием высокой температуры на поверхности хлеба происходит взаимодействие восстанавливающих сахаров с аминокислотами, в результате чего образуются различные карбонильные соединения и темноокрашенные продукты – меланоидины. Меланоидины и придают готовому продукту соответствующую окраску. Изменение цвета поверхности хлеба от светло-желтого до темно-коричневого происходит в интервале температур 130 –170°С, при температуре выше 170–175°С корка хлеба начинает обугливаться.
Качество хлеба оценивается по следующим показателям:
– внешний вид и цвет корки – корка должна быть румяная, поджаристая, не пригорелая, но и не бледная;
– кислотность – хлеб должен удовлетворять определенным нормам стандарта по кислотности;
– качество мякиша, структура пористости. Существует определенная шкала пористости, по которой определяют качество хлеба. Она должна быть доста
точно равномерной. Важным показателем качества хлеба является эластичность мякиша, его заминаемость. Если мякиш заминается или он недостаточно эластичен – хлеб плохой;
– влажность мякиша. Это очень важный показатель. Влажность мякиша должна соответствовать определенным нормам стандарта.
– объем хлеба – чем объем хлеба больше, тем он пористее, тем лучше пропитывается пищеварительными соками и, следовательно, лучше усваивается организмом. Для подового хлеба, то есть для хлеба, который выпекается не в форме, а на поду, большое значение имеет отношение высоты хлеба к его диаметру (Н/Д). Если мука плохая и хлеб получается низкий, то отношение высоты к диаметру будет очень низким;
– вкус и аромат хлеба – аромат хлеба зависит от очень сложного комплекса различных веществ, образующихся в процессе брожения теста и в процессе выпечки хлеба. Этот комплекс включает в себя различные альдегиды, органические кислоты и сложные эфиры. Особенно важную роль играют альдегиды, в первую очередь, фурфурол и оксиметилфурфурол.
Потенциометрический метод
Метод основан на нейтрализации кислот, содержащихся в продукте, раствором гидроокиси натрия до заранее заданного значения рН=8,9 с помощью блока автоматического титрования и индикации точки эквивалентности при помощи потенциометрического анализатора.
Аппаратура, материалы и реактивы: анализатор потенциометрический с диапазоном измерения 4-10 ед. рН с ценой деления шкалы 0,05 ед. рН.4; блок автоматического титрования, аппаратурно совместимый с потенциометрическим титратором и имеющий дозатор раствора (бюретку) вместимостью не менее 5 см с ценой деления не более 0,05 см; весы лабораторные 4-го класса точности с наибольшим пределом взвешивания 200 г; стаканы В-1-50 ТС, В-2-50 ТС, В-1-100 ТС, В-2-100 ТС; колбы 1-1000-2, 2-1000-2; пипетки 2-2-10, 2-2-20; цилиндры 1-50-1, 1-50-2, 3-50-1, 3-50-2; ступка фарфоровая с пестиком; натрия гидроокись, стандарт-титр по ТУ 6-09-2540, раствор с молярной концентрацией 0,1 моль/дм; вода дистиллированная.
Подготовка к измерениям
Подготовка приборов
Подключают блок автоматического титрования к анализатору согласно инструкции, прилагаемой к блоку. Затем подключают блок и анализатор к сети и прогревают их в течение 10 мин. Заполняют дозатор блока автоматического титрования раствором гидроокиси натрия.
Согласно инструкции, прилагаемой к потенциометрическому анализатору, настраивают его на такой диапазон измерения рН, который включил бы в себя рН=8,9.
Согласно инструкции, прилагаемой к блоку автоматического титрования, настраивают его на точку эквивалентности, равную 8,9 ед. рН, и устанавливают на блоке значение рН=4,0, начиная с которого подача гидроокиси натрия должна вестись по каплям. Устанавливают время выдержки после окончания титрования, равное 30 с.
Проведение измерений
1. Молоко, молокосодержащий продукт, молочный составной продукт, сливки, простокваша, ацидофилин, кефир, кумыс и другие кисломолочные продукты
2.В стакан вместимостью 50 см  отмеривают 20 см дистиллированной воды и 10 см анализируемого продукта. Смесь тщательно перемешивают.
отмеривают 20 см дистиллированной воды и 10 см анализируемого продукта. Смесь тщательно перемешивают.
При анализе сливок и кисломолочных продуктов переносят остатки продукта из пипетки в стакан путем промывания пипетки полученной смесью 3-4 раза.
3. В стакан помещают стержень магнитной мешалки и устанавливают стакан на магнитную мешалку. Включают двигатель мешалки и погружают электроды потенциометрического анализатора и сливную трубку дозатора блока автоматического титрования в стакан с продуктом. Включают кнопку "Пуск" блока автоматического титрования, а спустя 2-3 с, кнопку "Выдержка". Раствор гидроокиси натрия при этом начинает поступать из дозатора блока в стакан с продуктом, нейтрализуя последний. По достижении точки эквивалентности (рН=8,9) и истечении времени выдержки (30 с) процесс нейтрализации автоматически прекращается, а на панели блока автоматического титрования зажигается сигнал "Конец". После этого отключают все кнопки. Проводят отсчет количества раствора гидроокиси натрия, затраченного на нейтрализацию.
Мороженое, сметана
В стакане взвешивают 5 г продукта. Тщательно перемешивают продукт стеклянной палочкой, постепенно добавляют к нему 30 см воды и перемешивают. Проводят измерения.
Творог и творожные продукты
В фарфоровую ступку вносят 5 г продукта. Тщательно перемешивают и растирают продукт пестиком. Затем количественно переносят продукт в стакан вместимостью 100 см, смывая его небольшими порциями воды, нагретой до 35-40 °С. Общий объем воды равен 50 см. Затем смесь перемешивают и проводят измерения.
Обработка результатов
Кислотность в градусах Тернера находят умножением объема, см, раствора гидроокиси натрия, затраченного на нейтрализацию определенного объема продукта, на следующие коэффициенты:
10 - для молока, молочного составного продукта, сливок, простокваши, ацидофильного молока, кефира, кумыса и других кисломолочных продуктов;
20 - для мороженого, сметаны, творога и творожных изделий.
Предел допускаемой погрешности результата измерений при принятой доверительной вероятности  =0,95 составляет, °Т:
=0,95 составляет, °Т:
±0,8 - для молока, молочного составного продукта, сливок, мороженого;
±1,2 - для простокваши, ацидофильного молока, кефира, кумыса и других кисломолочных продуктов;
±2,3 - для сметаны;
±3,2 - для творога и творожных изделий.
Расхождение между двумя параллельными измерениями не должно превышать, °Т:
1,2 - для молока, молочного составного продукта, сливок, мороженого;
1,7 - для простокваши, ацидофильного молока, кефира, кумыса и других кисломолочных продуктов;
3,2 - для сметаны;
4,3 - для творога и творожных изделий.
За окончательный результат измерения принимают среднеарифметическое значение результатов двух параллельных определений, округляя результат до второго десятичного знака.
При большем расхождении испытание повторяют с четырьмя параллельными определениями. При этом расхождение между средним арифметическим значением результатов четырех определений и любым значением из четырех результатов определения не должно превышать, °Т:
0,8 - для молока, молочного составного продукта, сливок, мороженого;
1,2 - для простокваши, ацидофильного молока, кефира, кумыса и других кисломолочных продуктов;
2,3 - для сметаны;
3,2 - для творога и творожных изделий.
При большем расхождении приготовляют заново все реактивы, проводят государственную поверку используемых приборов и повторяют испытание с четырьмя параллельными определениями. В этом случае при наличии расхождения, больше вышеуказанных значений, выполнение данной работы поручают оператору более высокой квалификации.
Метод с применением индикатора фенолфталеина
Метод основан на нейтрализации кислот, содержащихся в продукте, раствором гидроокиси натрия в присутствии индикатора фенолфталеина.
Аппаратура, материалы и реактивы: весы лабораторные 4-го класса точности с наибольшим пределом взвешивания 200 г; центрифуга по ТУ 27-32-26-77; шкаф сушильный с терморегулятором, позволяющий поддерживать температуру (50±5) °С; баня водяная; термометр ртутный стеклянный с диапазоном измерения 0-100 °С и ценой деления 0,1°С; колбы 1-100-2, 2-100-2, 1-1000-2, 2-1000-2; колбы П-2-50-34 ТС, П-2-100-34 ТС, П-2-250-34 ТС, П-2-250-50; стаканы В-1-100 ТС, В-1-250 ТС; воронки В-36-80 ХС; жиромеры стеклянные 1-40; 2-0,5; пипетки 1-2-1, 2-2-1, 4-2-1, 2-2-5, 2-2-10, 2-2-20; цилиндр 1-1-100; бюретки 6-1-10-0,02, 6-2-10-0,02, 7-1-10-0,02, 7-2-10-0,02; ступка фарфоровая с пестиком; палочки стеклянные; штатив лабораторный; пробки для жиромеров; бумага фильтровальная; натрия гидроокись стандарт-титр по ТУ 6-09-2540 раствор молярной концентрации 0,1 моль/дм; фенолфталеин по ТУ 6-09-5360, 70%-ный спиртовой раствор массовой концентрации фенолфталеина 10 г/дм; кобальт сернокислый, раствор массовой концентрации сернокислого кобальта 25 г/дм; эфир диэтиловый для наркоза; вода дистиллированная; спирт этиловый ректификованный; спирт этиловый технический (гидролизный)или спирт этиловый ректификованный технический.
Подготовка к анализу
1. Приготовление контрольных эталонов окраски для молока и сливок
В колбу вместимостью в 100 или 250 см отмеривают молоко или сливки и дистиллированную воду объемах, указанных в табл.1, и 1 см раствора сернокислого кобальта. Смесь тщательно перемешивают.
| Наименование продукта | Объем
продукта, см
| Объем дистиллированной воды, см
|
| Молоко, молокосодержащий продукт | 10 | 20 |
| Молочный составной продукт | 10 | 40 |
| Сливки | 10 | 20 |
| Простокваша, ацидофилин, кефир, кумыс и другие кисломолочные продукты | 10 | 20 |
Срок хранения эталона не более 8 ч при комнатной температуре.
Приготовление контрольных эталонов окраски для смеси этилового спирта и диэтилового эфира
К 10 см спирта добавляют 10 см диэтилового эфира и 1 см раствора сернокислого кобальта. Смесь тщательно перемешивают.
Приготовление контрольных эталонов окраски для сливочного масла и масляной пасты, их жировой фазы
К 5 г масла, расплавленного, как указано в п.3.2.6, добавляют 20 см нейтрализованной смеси спирта и эфира и 1 см раствора сернокислого кобальта. Смесь перемешивают.
Приготовление контрольных эталонов окраски для плазмы сливочного масла и масляной пасты
К 10 см плазмы, приготовленной как указано в п.3.2.7, добавляют 20 см воды. Полученной смесью 3-4 раза промывают пипетку и добавляют 1 см раствора сернокислого кобальта. Смесь перемешивают.
Приготовление смеси этилового спирта и диэтилового эфира
Смесь этилового спирта и диэтилового эфира готовят непосредственно перед измерением кислотности сливочного масла и масляной пасты или его жировой фазы следующим образом.
В колбу вместимостью 50 см приливают по 10 см спирта и эфира, 3 капли фенолфталеина и нейтрализуют смесь раствором щелочи до появления слабо-розового окрашивания, не исчезающего в течение 1 мин и соответствующего контрольному эталону окраски.
Приготовление жировой фазы сливочного масла и масляной пасты
В сухой чистый стакан вместимостью 250 см отвешивают около 150 г исследуемого масла. Стакан помещают в водяную баню или сушильный шкаф при температуре (50±5) °С и выдерживают до полного расплавления и разделения масла на жир и плазму. Стакан вынимают из водяной бани (сушильного шкафа) и осторожно сливают верхний слой жира, фильтруя его через бумажный фильтр в колбу вместимостью 250 см.
Приготовление плазмы сливочного масла и масляной пасты
Оставшуюся в стакане плазму переносят в жиромер 2-0,5. Жиромер плотно закрывают пробкой, помещают в центрифугу и центрифугируют 5 мин с частотой вращения 1000 мин. Затем жиромер помещают в стакан с холодной водой градуированной частью вверх и выдерживают до застывания молочного жира, отделившегося от плазмы в процессе центрифугирования. Свободную от жира плазму осторожно выливают в сухой чистый стакан вместимостью 100 см и тщательно перемешивают стеклянной палочкой.
Проведение анализа
1. Молоко, молокосодержащий продукт, молочный составной продукт, сливки, простокваша, ацидофилин, кефир, кумыс и другие кисломолочные продукты.
2. В колбу вместимостью 100 до 250 см отмеривают дистиллированную воду и анализируемый продукт в объемах, указанных в табл.1, и три капли фенолфталеина. При анализе сливок и кисломолочных продуктов переносят остатки продукта из пипетки в колбу путем промывания пипетки полученной смесью 3-4 раза.
3. Смесь тщательно перемешивают и титруют раствором гидроокиси натрия до появления слаборозового окрашивания, для молока и сливок, соответствующего контрольному эталону окраски по п.3.2.1, не исчезающего в течение 1 мин.
Для молочного составного продукта для более точного установления конца титрования рядом с титруемой пробой ставят контрольную колбу с 10 см той же пробы молока и 40 см дистиллированной воды.
Мороженое, сметана
В неокрашенном мороженом и сметане кислотность определяют следующим образом: в колбе вместимостью 100 или 250 см отвешивают 5 г продукта, добавляют 30 см воды и три капли фенолфталеина. Смесь тщательно перемешивают и титруют раствором гидроокиси натрия до появления слабо-розового окрашивания, не исчезающего в течение 1 мин.
Кислотность окрашенного мороженого определяют следующим образом: отвешивают в колбе вместимостью 250 см 5 г мороженого, добавляют 80 см воды и три капли фенолфталеина. Смесь тщательно перемешивают и титруют раствором щелочи до появления слабо-розового окрашивания, не исчезающего в течение 1 мин.
Для определения конца титрования окрашенного мороженого колбу с титруемой смесью помещают на белый лист бумаги и рядом помещают колбу со смесью: 5 г данного образца мороженого и 80 см воды.
Творог и творожные продукты
В фарфоровую ступку вносят 5 г продукта. Тщательно перемешивают и растирают продукт пестиком. Затем прибавляют небольшими порциями 50 см воды, нагретой до температуры 35-40 °С и три капли фенолфталеина. Смесь перемешивают и титруют раствором щелочи до появления слабо-розового окрашивания, не исчезающего в течение 1 мин.
Масло сливочное и масляная паста, их жировая фаза, плазма
Определение кислотности сливочного масла и масляной пасты
В колбе вместимостью 50 и 100 см отвешивают 5 г сливочного масла и масляной пасты, нагревают колбу в водяной бане или сушильном шкафу при температуре (50±5) °С до расплавления масла, вносят 20 см нейтрализованной смеси спирта с эфиром, три капли фенолфталеина и титруют раствором щелочи при постоянном перемешивании до появления слабо-розового окрашивания, не исчезающего в течение 1 мин и соответствующего контрольному эталону окраски.
Определение кислотности жировой фазы сливочного масла и масляной
пасты
В колбе вместимостью 50 или 100 см взвешивают 5 г жира. Затем проводят аналогичный анализ.
Определение кислотности плазмы сливочного масла и масляной пасты
В плоскодонную колбу вместимостью 100 см приливают 10 см плазмы, 20 см дистиллированной воды. Полученной смесью 3-4 раза промывают пипетку, затем прибавляют 3 капли фенолфталеина и титруют при постоянном перемешивании раствором щелочи до появления слабо-розового окрашивания, не исчезающего в течение 1 мин и соответствующего контрольному эталону окраски.
Обработка результатов
Кислотность, в градусах Тернера (°Т), находят умножением объема, см, раствора гидроокиси натрия, затраченного на нейтрализацию кислот, содержащихся в определенном объеме продукта, на следующие коэффициенты:
10 - для молока, молочного составного продукта, сливок, простокваши, ацидофильного молока, кефира, кумыса, других кисломолочных продуктов, а также плазмы сливочного масла и масляной пасты;
20 - для мороженого, сметаны, творога и творожных изделий.
Кислотность сливочного масла и масляной пасты и их жировой фазы в градусах Кеттстофера (°К) находят умножением на два объема раствора гидроокиси натрия, затраченного на нейтрализацию кислот, содержащихся в 5 г продукта.
Допускаемая погрешность результата анализа при принятой доверительной вероятности =0,95, составляет:
±1,9 °Т - для молока, молочного составного продукта, сливок, простокваши, ацидофильного молока, кефира, кумыса, других кисломолочных продуктов и мороженого;
±2,3 °Т - для сметаны;
±3,6 °Т - для творога и творожных изделий;
±0,1 °К - для масла сливочного и масляной пасты и их жировой фазы;
±0,5 °Т - для плазмы сливочного масла и масляной пасты.
Расхождение между двумя параллельными определениями не должно превышать:
2,6 °Т - для молока, молочного составного продукта, сливок, простокваши, ацидофильного молока, кефира, кумыса, других кисломолочных продуктов и мороженого;
3,2 °Т - для сметаны;
5,0 °Т - для творога и творожных изделий;
0,1 °К - для масла сливочного и масляной пасты и их жировой фазы;
0,6 °Т - для плазмы сливочного масла и масляной пасты.
За окончательный результат анализа принимают среднеарифметическое значение результатов двух параллельных определений, округляя результат до второго десятичного знака.
При большем расхождении испытание повторяют с четырьмя параллельными определениями. При этом расхождение между средним арифметическим значением результатов четырех определений и любым значением из четырех результатов определения не должно превышать:
1,8 °Т - для молока, молочного составного продукта, сливок, простокваши, ацидофильного молока, кефира, кумыса, других кисломолочных продуктов и мороженого;
2,3 °Т - для сметаны;
3,6 °Т - для творога и творожных изделий;
0,1 °К - для масла сливочного и масляной пасты и их жировой фазы;
0,5 °Т - для плазмы сливочного масла и масляной пасты.
При большем расхождении приготовляют заново все реактивы, проводят государственную поверку используемых приборов и повторяют испытание с четырьмя параллельными определениями. В этом случае при наличии расхождения больше вышеуказанных значений выполнение данной работы поручают оператору более высокой квалификации
Контрольные вопросы:
1. Требования, предъявляемые к молоку в сыроделии
2. Подготовка молока к свертыванию
3. Свертывание белков молока и получение сырной массы
Список литературы
а) основная литература:
1. Новиков Н.Н. Биохимия растений / Н.Н. Новиков. – М.: КолосС, 2010 г. – 679 с.
2. Новиков Н.Н. Лабораторный практикум по биохимии растений: Учебное пособие / Н.Н. Новиков, Т.В. Таразанова. – М.: издательство РГАУ-МСХА имени К.А.Тимирязева, 2012 г. – 97 с.
3. Филипцова Г. Г. Биохимия растений: методические рекомендации к лабораторным занятиям, задания для самост. работы студентов / Г. Г. Филипцова, И. И. Смолич.– Мн.: БГУ, 2004 г.– 60 с.
б) дополнительная литература:
1. Берёзов Т.Т. Биологическая химия / Т.Т. Берёзов, Б.Ф. Коровкин. – М.: Медицина, 2002 г. – 528 с.
2. Дука М. Физиология растений: практикум для студентов биолого-почвенного факультета / М. Дука, Т. Хомченко, Е. Савка. – Кишинау, 2003 г. – 133 с.
3. Запромётов М.Н. Фенольные соединения и их роль в жизни растений /М.Н. Запромётов. – М.: Наука, 1996 г. – 45 с.
4. Казаков Е.Д. Биохимия зерна и хлебопродуктов / Е.Д. Казаков, Г.П. Карпиленко. – СПб.: Гиорд, 2005 г.– 510 с.
5. Кислухина О.В. Витаминные комплексы из растительного сырья / О.В. Кислухина. – М.: ДеЛи принт, 2004 г.– 308 с.
6. Практикум по биохимии / под ред. А. А. Чиркина. – Мн.: 2002 г. – 512 с.
7. Руководство к практическим занятиям по биохимии / под ред. Е. С. Се-верина. – М.: Медицина, 2000 г. – 126 с.
8. Румянцев Е.В. Химические основы жизни / Е.В. Румянцев, Е.В. Антина, Ю.В. Чистяков. – М.: КолосС, 2007 г.– 560 с.
9. Татарченко И.И. Химия субтропических и пищевкусовых продуктов / И.И. Татарченко, И.Г. Мохначёв, Г.И. Касьянов. – М.: Академия, 2003 г.– 256 с.
10. Щербаков В.Г. Биохимия и товароведение масличного сырья / В.Г. Щербаков, В.П. Лобанов. – М.: КолосС, 2003 г.– 360 с.
СОДЕРЖАНИЕ
| стр. | |
| Введение……………………………………………………………………. | 3 |
| Модуль 1. Характеристика и биологические функции органических соединений…………………. Тема 1. Введение в биохимию……………………………………………. Лабораторная работа № 1. Правила работы и техника безопасности в химической лаборатории…………………………………………………... Контрольные вопросы………………………………………………………. | 3 3 3 5 |
| Тема 2. Состав, строение и биологические функции углеводов и липидов……………………………………………………………………. Лабораторная работа № 2. Особенности сахаров и методов их определения. Определение редуцирующих сахаров и сахарозы………………… Контрольные вопросы………………………………………………………. | 5 5 9 |
| Лабораторная работа №3. Определение кислотного, йодного и перекисного числа растительных жиров……………..……………………….... Контрольные вопросы ……………………………………………………… | 9 13 |
| Тема 3. Состав, строение и биологические функции белков и витаминов. Качественные реакции на белки………………………….. Лабораторная работа № 4. Определение белков по методу Лоури…… Колориметрическое определение аскорбиновой кислоты в растительных продуктах…………..………………………….……….… Определение витаминов В1 и В2 в растениях……………………………… Контрольные вопросы………………………………………………………. | 13 13 17 18 23 |
| Тема 4. Ферменты. Состав, строение и биологические функции……. Лабораторная работа № 5. Определение активности пероксидазы…… Определение активности каталазы………………………………………… Определение активности аскорбатоксидазы……………………………… Определение активности тирозиназы…………………………………….. Определение гидролитической активности липазы…………………..…. Контрольные вопросы…………………………………………………….. | 24 26 27 28 30 30 32 |
| Модуль 2. Особенности обменных процессов органических соединений……..…………………………. Тема 6. Обмен углеводов, липидов и азотистых веществ в организмах……………………………………………………………….. Лабораторная работа № 6. Определение амминного азота формольным титрованием…….…………………………………….…..….. Определение нитратного азота…………………………………………….. Контрольные вопросы…………………………………………………….. | 33 33 33 35 44 |
| Тема 7. Органические кислоты и вещества вторичного происхождения……………………………………………………………. Лабораторная работа № 7. Определение органических кислот и кислых солей в растительном материале……………………………….…… Определение общей кислотности………………………………….……… Контрольные вопросы……………………………………………………… | 45 46 48 49 |
| Тема 8. Биохимия растительных продуктов…………………………… Лабораторная работа № 8. Определение кислотности зерна.………….. Анализ качества полученного хлеба………………………………….…… Контрольные вопросы……………………………………………………… | 49 49 55 56 |
| Тема 9. Биохимия молока и мяса………………………………………… Лабораторная работа № 9. Определение кислотности молока………. Контрольные вопросы……………………………………………………… | 56 56 66 |
| Список литературы……………………………………………………….. | 67 |
Методические указания для проведения лабораторных занятий
по курсу «Биохимия сельскохозяйственной продукции»
(для бакалавров технологического факультета по направлению: 35.03.07 –
Технология производства и переработки сельскохозяйственной
продукции)
Тверь, 2017
Составитель: к. с.-х. н., доцент кафедры агрохимии и земледелия
Шилова О.В.
Рецензент: к. б. н., доцент кафедры биологии животных, зоотехнии и основ
ветеринарии Тверской ГСХА Корецкая Е.А.
Шилова О.В. Методические указания для проведения лабораторных занятий по курсу «Биохимия сельскохозяйственной продукции» (для бакалавров технологического факультета по направлению: 35.03.07 – Технология производства и переработки сельскохозяйственной продукции) – Тверь, 2017– 69 с.
Председатель методической комиссии Акимов А.А.
Введение
Цели дисциплины: формирование современных представлений, знаний и умений о превращениях веществ и энергии в живых организмах, химическом составе сельскохозяйственной продукции растительного и животного происхождения, биохимических процессах, происходящих в ней при хранении и переработке.
Задачи дисциплины: изучение строения и биологических функций важнейших органических веществ; механизмов ферментативных и биоэнергетических превращений в организмах; химического состава сельскохозяйственной продукции и биохимических процессов, происходящих в ней при хранении и переработке;
– оценка качества и технологических свойств сельскохозяйственной продукции по биохимическим показателям;
– применение знаний о химическом составе и биохимических процессах при обосновании технологий производства, хранения и переработки сельскохозяйственной продукции;
– ознакомление с современными методами и достижениями биохимической науки.
Дата: 2018-12-21, просмотров: 1246.