В литературе описано более 60 случаев изодисомии (материнская дупликация) и гетеродисомии материнского происхождения. На хромосоме 7 расположены три района, содержащие импринтированные гены. Район короткого плеча 7(р11.2-р13) содержит импринтированный ген GRB10, кодирующий цитоплазматический адаптерный белок, который служит негативным регулятором сигналинга тирозинкиназных рецепторов (например, IGF1R). Мышиный ген импринтирован, экспрессируется с материнской хромосомы во всех тканях, кроме мозга, в котором экспрессируется отцовский аллель. Делеции материнского аллеля гена приводят к увеличению роста у потомства, свидетельствуя о его функционировании в качестве негативного регулятора роста. У человека отцовская экспрессия гена установлена в головном и спинном мозге, материнская - в скелетных мышцах, а во всех остальных тканях ген экспрессируется биаллельно. Поиск мутаций в гене GRB10 у больных с СРС не привел к находкам, как, впрочем, и попытка обнаружения аномалий метилирования в регуляторной области.
Длинное плечо хромосомы 7q32.2 содержит пять импринтированных генов, включая MEST, COPG2IT1 и MESTIT1, экспрессирующихся с отцовской хромосомы, и CPA4 и KLF14 (экспрессируются с материнской хромосомы).
MEST имеет две изоформы, одна из которых экспрессируется с отцовского аллеля, а вторая, использующая альтернативный первый экзон, - биаллельно во всех тканях, кроме плаценты. Нокаут гена у мышей приводит к малому размеру потомства, но ни мутаций, ни аномального метилирования у пациентов с СРС не обнаружено. Импринтированная антисмысловая РНК MESTIT1, исследованная на предмет мутаций, таковых не показала. Они также не были обнаружены в генах CPA4 и KLF14.
Импринтированный локус на хромосоме 7q21.3 содержит гены PEG10 и SGCE, имеющие отцовскую экспрессию. PPP1R9A экспрессируется с материнского аллеля в эмбриональных скелетных мышцах и экстраэмбриональных тканях, а ген TFP12 - с материнского аллеля в плаценте. Мутации гена SGCE вызывают миоклональную дистонию, а делеции PEG10 у мышей приводят к ранней эмбриональной гибели. У пациентов с СРС структурные нарушения в указанных импринтированных генах не обнаружены. Именно поэтому на сегодняшний день сложно сказать, какой именно ген на хромосоме 7 вносит решающий вклад в формирование фенотипа СРС.
Аномалии импринтинга гена Н19 при синдроме Сильвера-Рассела.
Хромосомный район 11р15.5 содержит протяженный кластер импринтированных генов, вовлеченных в патогенез СБВ. До тех пор пока не было обнаружено двух материнских дупликаций этого хромосомного района у больных, никто не предполагал, что его изменения могут вызывать развитие еще одного синдрома. Тщательный молекулярный анализ показал, что ряд пациентов имеет гипометилирование или потерю импринтинга гена Н19, имеющего материнскую экспрессию, т.е. ген начинает экспрессироваться и с отцовского аллеля. Это связано с тем, что ЦИ1 на отцовской хромосоме не подвергается метилированию и связывает белок CTCF, что приводит к биаллельной экспрессии Н19 и биаллельной инактивации IGF2 (рис. 16). В частности, показано, что ДМР в 5’-районе IGF2 на отцовской хромосоме также гипометилирован. Второй центр импринтинга (KCNQ1OT1), расположенный в гене KCNQ1, за редким исключением, не подвергается изменениям метилирования у больных с СРС.
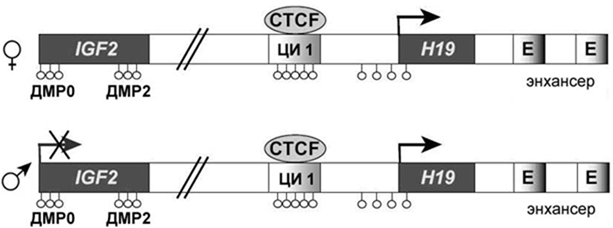
Рисунок 16. Биаллельная экспрессия гена Н19 при СРС.
В клетках пациентов с СРС аномалии метилирования Н19 регистрируют с частотой 0-35%. Это означает, что только часть клеток теряет метилирование Н19 на отцовской хромосоме. Мозаичная этиология аномалий импринтинга показывает, что нарушения возникают на уровне первых делений дробления, а значит, основная масса случаев заболевания имеет спорадический характер.
В 40% случаев СРС, где молекулярная патология не выявлена, могут быть другой, еще не установленный молекулярный дефект и сложности клинической диагностики синдрома.
ОДНОРОДИТЕЛЬСКАЯ ДИСОМИЯ ПО ХРОМОСОМЕ 14
Район хромосомы 14q32.2 (рис. 17) содержит кластер импринтированных генов: часть экспрессируется с отцовской хромосомы – DLK1, RTL1 и DIO3, а другие – гены нкРНК GTL2, RTL1as, MEG3, MEG8, MEG9, snoРНК и miРНК – с материнской. DLK1 - регулирует дифференцировку преадипоцитов, экспрессируется в нейроэндокринных тканях, особенно в корковом слое надпочечников. RTL1 - ретротранспозон-подобный ген, экспрессируется в плаценте и позднем фетальном периоде. DIO3 - йодтиронин дейодиназа 3 типа имеет несколько транскриптов: один, протяженностью 2,1 т.н. экспрессируется в плаценте, фетальной печени и матке, другой – 3,2 т.н. – в яичках, мочевом пузыре и матке, третий – 4,8 т.н. – в сердце и скелетных мышцах. Функция нкРНК GTL2 неизвестна, но показано, что интронный ДМР содержит сайт связывания белка CTCF, что предопределяет ее регуляторные функции. Функции RTL1as, MEG8 и MEG9 неизвестны. Паттерны экспрессии генов, зависящие от родительского происхождения, регулируются первичным межгенным ДМР, имеющим герминальное происхождение (MEG3/DLK1: IG-ДМР), и вторичным ДМР, функционирующим уже после оплодотворения (MEG3-ДМР). Оба ДМР метилированы на отцовской хромосоме в норме.
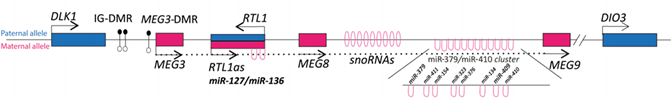
Рисунок 17. Структурная организация импринтированного района хромосомы 14. Синим цветом обозначены последовательности, которые имеют отцовскую экспрессию, розовым – материнскую; темные и светлые овалы – ДМР.
Синдром Темпл (СТ) (OMIM #616222) характеризуется пренатальной и постнатальной задержкой роста и ранним началом полового созревания, малым ростом, гипотонией, трудностями кормления в раннем детстве, моторной задержкой, слабостью суставов, ожирением нижней части туловища и признаками дисморфогенеза, такими как широкий лоб и короткий нос с широким кончиком и микроакрией. В связи с относительно легкими и зависимыми от возраста фенотипическими проявлениями популяционная частота синдрома неизвестна, и, вполне вероятно, заболевание сложно однозначно диагностировать. Кроме того, СТ имеет несколько общих клинических признаков, характерных для СПВ и СРС.
Синдром обусловлен изменением экспрессии импринтированных генов на хромосоме 14q32.2. Механизмами, приводящими к функциональной гемизиготности импринтированных генов 14q32, и клиническим фенотипическим аномалиям, являются: 1) материнская ОРД по хромосоме 14 (78%); 2) изолированная потеря метилирования в районе MEG3-ДМР (12%); 3) делеции 14q32 отцовского происхождения (10%).
Характерными признаками синдрома Кагами-Огата (СКО) (OMIM #608149) являются: крупные размеры плода (вес при рождении непропорционально больше, чем длина), многоводие, плацентомегалия, сниженный/отсутствующий сосательный рефлекс и гиповентиляция в неонатальном периоде, дефекты брюшной стенки, начиная от омфалоцеле до диастаза прямой кишки, специфические признаки дисэмбриогенеза (полные щеки, вдавленная переносица, микрогнатия, короткая складчатая шея и выступающий фильтр), маленькая колоколообразная грудная клетка (патогномоничный признак) с ребрами, похожими на одежную вешалку и различной степени выраженности задержка развития и умственная отсталость. Некоторые неспецифические фенотипические признаки сходны с таковыми при СБВ. Синдром ассоциирован с повышенным риском развития гепатобластомы (9%) и неонатальной смертностью до 20-25%.
СКО может быть вызван тремя различными молекулярными событиями: 1) отцовская ОРД по хромосоме 14 (65% случаев); 2) микроделеции, повреждающие импринтированный район 14q32.2 на материнской хромосоме (20%); 3) гиперметилирование ДМР MEG3 на материнской хромосоме (15%). Если отцовская ОРД 14 и гиперметилирование MEG3 возникают спорадически, то микроделеции могут привести к наследуемому по материнской линии СКО. Недавно было показано, что делеции импринтированного района не обязательно включают ДМР, поэтому нормальный паттерн метилирования не исключает наличие синдрома.
Было показано, что гипометилирование в домене DLK1/MEG3 приводит к снижению экспрессии импринтированных генов, таких как IGF2, SNURF и IPW, а также ряда других неимпринтированных генов, участвующих в стимулировании роста. Изменения в экспрессии могут отражать, прямо или косвенно, участие нкРНК MEG3 и MEG8 района 14q32.2 в этом процессе. Было показано, что нкРНК регулируют экспрессию генов как in cis, так и in trans посредством ассоциации с модификаторами хроматина. Гиперэкспрессия или инактивация экспрессии MEG3 и MEG8 в культурах нормальных фибробластов может быть связана с нарушением регуляции импринтированных генов в хромосомных районах 11p15.5 и 15q11-q13, в частности, установлено: 1) гиперэкспрессия MEG3 связана со снижением уровня транскриптов IPW; 2) гиперэкспрессия MEG8 связана с более низкими уровнями SNURF и 3) совместная гиперэкспрессия MEG3 и MEG8 связана с более низкими уровнями транскриптов IGF2. Это показывает, что MEG3 и MEG8 могут регулировать in trans экспрессию других импринтированных генов. Ранее было установлено, что гиперэкспрессия IPW может подавлять экспрессию MEG3. Таким образом, вполне вероятно, что в эпитранскриптоме может существовать система реципрокного контроля, которая связывает работу нкРНК импринтированных районов, а это, в свою очередь, может способствовать клиническому перекрыванию фенотипических проявлений СПВ, СБВ и СРС.
ПСЕВДОГИПОПАРАТИРЕОИДИЗМ И ИМПРИНТИРОВАННЫЙ ЛОКУС ХРОМОСОМЫ 20q13.2
Псевдогипопаратиреоидизм (ПГПТ) - гетерогенная группа эндокринных нарушений, характеризующаяся почечной резистентностью к паратиреоидному гормону (ПТГ), вызывающей гипокальциемию, гиперфосфатемию и повышенный уровень циркулирующего ПТГ. В зависимости от молекулярной патологии ПГПТ включает другие эндокринные патологии, связанные с резистентностью к воздействию гормонов и рядом не эндокринных особенностей. В целом распространенность ПГПТ оценивается, как 1,1 на 100 000. Клинически гетерогенные фенотипы являются результатом структурных и функциональных изменений гена GNAS, кодирующего α-субъединицу белка, связывающего гуанин (Gsα).
Псевдогипопаратиреоз типа 1A (ПГПТ1А) (OMIM #103580) обусловлен мутациями с потерей функции в материнском аллеле гена GNAS. У пациентов с ПГПТ1A наблюдается генерализованная гормонорезистентность различной степени, умственная отсталость, ожирение, связанное со снижением энергетических затрат в состоянии покоя, наследственная остеодистрофия Олбрайт (АНО), проявляющаяся низкорослостью, округлым лицом, подкожными оссификатами, брахидактилией и другими скелетными аномалиями.
Потеря функции Gsα на отцовском аллеле вызывает псевдопсевдогипопаратиреоз (ППГПТ) (OMIM #612463). Поскольку почечные тубулярные клетки преимущественно экспрессируют материнский аллель GNAS, то мутация, унаследованная от отца, приводит к нормальному почечному ответу на ПТГ, вызывая АНО без сопутствующих эндокринных нарушений. Отцовские мутации с потерей функции могут приводить к прогрессирующей костной гетероплазии (ПКГ) (OMIM#166350), состоянию, характеризующемуся подкожной оссификацией, появляющейся в детском возрасте и прогрессирующей с возрастом, с вовлечением подкожных и более глубоких соединительных тканей, хотя АНО или гормонорезистентность отсутствуют. Пациенты с ППГПТ и ПКГ имеют вдвое сниженную экспрессию Gsα в эритроцитах, хотя в норме GNAS экспрессируется биаллельно. AHO может быть вызвана гаплонедостаточностью Gsα в тканях, где GNAS экспрессируется с обоих аллелей.
Напротив, псевдогипопаратиреоз типа 1B (ПГПТ1B) (OMIM #603233) клинически характеризуется изолированной почечной резистентностью к ПТГ и в некоторых случаях резистентностью к ТТГ. У таких пациентов редко обнаруживается фенотип АНО. Интересно, что экспрессия Gsα в эритроцитах умеренно снижается у пациентов с АНО. Все пациенты с ПГПТ1B имеют, по крайней мере, потерю метилирования в ДМР GNAS A/B, что, вероятно, приводит к снижению экспрессии GNAS-Gsα транскрипта в тканях, подверженных импринтингу. Гормональная резистентность обусловлена потерей метилирования материнского аллеля. В целом, 20% случаев ПГПТ1B наследуются и вызываются делециями в центрах импринтинга, в то время как остальные 80% являются спорадическими и связаны с аномалиями метилирования, охватывающими весь локус GNAS. Небольшое количество спорадических случаев ПГПТ1B связано с отцовской ОРД хромосомы 20q. Дупликации и делеции в локусе GNAS были выявлены всего у нескольких пациентов, а в большинстве случаев этиологию заболевания установить не удается. Описаны как спорадические, так и семейные случаи заболевания, причем последние наследуются по аутосомно-доминантному типу с неполной пенетрантностью. Анализ больших семей показал, что резистентность к паратиреоидному гормону развивается только в том случае, если дефект наследуется по материнской линии, т.е. тип наследования подобен тому, который обнаруживают в семьях с ПГПТ1А/ППГПТ.
Материнская ОРД по хромосоме 20, как правило, вызывается редукцией трисомии, произошедшей в результате нерасхождения во время второго деления мейоза, до дисомии. Пациенты имеют маленький рост, экстремальные сложности вскармливания, вызванные невозможностью сосания в первые годы жизни. Фенотипические признаки пациентов с материнской ОРД перекрываются с таковыми при СРС и требуют дифференциальной диагностики.
Ген картирован на хромосоме 20q13.2. Локус GNAS имеет три альтернативных первых экзона (А/B, XL и NESP55), которые сплайсируются с экзонами 2-13. Это приводит к возникновению различных транскриптов (рис. 18). В непосредственной близости от альтернативных экзонов расположены ДМР, что приводит к тому, что NESP55 экспрессируется только с материнской хромосомы, хотя XL, экзон А/В и антисмысловой транскрипт NESP55АС экспрессируются с отцовской хромосомы.
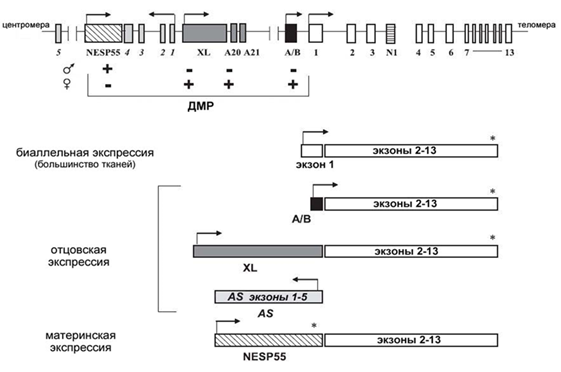
Рисунок 18. Экзон-интронная организация локуса GNAS и транскрипты, получаемые в результате альтернативного сплайсинга: Gsα-транскрипт экспрессируется биаллельно, за исключением проксимальных почечных канальцев, щитовидной железы, гонад и гипоталамуса. XL, A/B и AS (антисмысловой транскрипт) имеют отцовскую экспрессию, а NESP55 - материнскую. Промоторные районы указанных транскриптов имеют дифференциальное метилирование на отцовской (р+/-) и материнской хромосоме (м+/-). Звездочкой отмечены терминирующие кодоны. ДМР - дифференциально метилированные районы.
Более протяженный вариант белка Gsα - XL - экспрессируется преимущественно в нейроэндокринных тканях и нервной системе. Оба белка различаются только по N-концу. Другой белок гена GNAS - NESP55 - хромогранинподобный нейроэндокринный секреторный белок. Так же как и в случае XL, альтернативный экзон сплайсируется с экзонами 2-13, но в результате существования в нем терминирующего кодона NESP55 не имеет гомологии с белком Gsα. Указанные транскрипты экспрессируются с отцовской или материнской хромосомы соответственно. Два других транскрипта - A/B и антисмысловой NESPAS, у которого есть свои собственные экзоны, - экспрессируются во всех тканях с отцовской хромосомы, но не транслируются. Промотор GNAS, кодирующий Gsα, не имеет дифференциального метилирования и экспрессируется биаллельно практически во всех тканях, экспрессия с материнского аллеля происходит только в проксимальных канальцах почек, щитовидной железе, гипофизе и гонадах.
Если у пациентов с ПГПТ1А, как правило, обнаруживают мутации в GNAS, то у пациентов с ПГПТ1В таковых не обнаружено. В то же время у последних определяют потерю метилирования ДМР локуса GNAS, особенно в области альтернативного экзона А/В, что приводит к биаллельной экспрессии А/В-транскрипта (рис. 19).
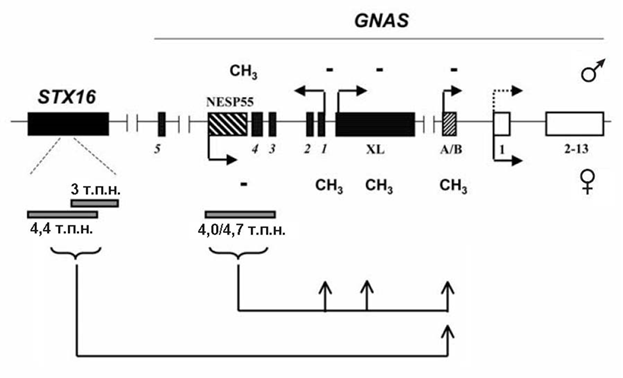
Рисунок 19. Структурная молекулярная патология, которая приводит к нарушению функционирования импринтированного локуса GNAS. Делеция 3 т.п.н. гена STX16 нарушает элемент, устанавливающий или поддерживающий метилирование ДМР А/В. Делеции ДМР NESP55 повреждают элемент, контролирующий импринтинг всего локуса GNAS на материнской хромосоме. СН3 – наличие метилирования.
Гетерозиготная делеция протяженностью 3 тыс. пар нуклеотидов обнаружена в 220 тыс. пар нуклеотидов центромернее экзона А/В как у пациентов со спорадическим, так и унаследованным от матери ПГПТ1В. Эта делеция повреждает экзоны 4-6 гена STX16, кодирующего белок синтаксин 16, который вовлечен в межклеточные взаимодействия. Другой вариант делеции повреждает экзоны 2-4, т.е. наименьший район перекрывания делеций расположен в районе экзона 4 и содержит CpG-обогащенный участок, который не подвержен дифференциальному метилированию. Очевидно, что этот ген не может быть вовлечен в патогенез заболевания, но все пациенты с делецией имеют потерю метилирования в районе экзона А/В; она нарушает cis-действующий элемент, контролирующий импринтинг, который устанавливает и/или поддерживает метилированный статус ДМР экзона А/В на материнской хромосоме.
В нескольких семьях с заболеванием и аномалиями импринтинга локуса GNAS была обнаружена делеция ДМР экзона NESP55, которая одновременно повреждала экзоны 3 и 4 антисмыслового транскрипта. Материнские делеции ДМР NESP55 полностью уничтожают материнский импринт локуса GNAS, приводя к биаллельной экспрессии XL, A/B и антисмыслового транскрипта. В этом районе содержится еще один регуляторный элемент, необходимый для полноценного метилирования материнского аллеля GNAS.
В основной массе спорадических случаев ПГПТ1В, сопровождающихся нарушением импринтинга как ДМР экзона А/В, так и всех ДМР локуса GNAS, но при отсутствии делеций в районе STX16 и ДМР NESP55, можно предполагать молекулярные нарушения в других отдаленных регуляторных районах, которые еще требуется определить. Все молекулярные изменения при ПГПТ1В повреждают импринтинг экзона А/В. Поскольку при заболевании нарушается экспрессия Gsα, правильное метилирование на материнской хромосоме экзона А/В, располагающегося в непосредственной близости от промотора Gsα, - необходимое условие экспрессии этого белка, по крайней мере, в проксимальных почечных канальцах.
Несмотря на то что все молекулярно-генетические причины, приводящие к ПГПТ1В, еще не выяснены, молекулярная диагностика может быть основана на анализе метилирования всех ДМР локуса, а ДМР экзона А/В - в первую очередь. Исследование можно проводить с использованием метилчувствительной ПЦР (подобно анализу потери метилирования гена IGF2 при СБВ) и метилспецифической ПЦР.
ТРАНЗИТОРНЫЙ НЕОНАТАЛЬНЫЙ САХАРНЫЙ ДИАБЕТ
Транзиторный неонатальный сахарный диабет (ТНСД) (#OMIM 601 410) - редкое заболевание (частота - 1:500 000 новорожденных), которое манифестирует в раннем неонатальном периоде гипергликемией, глюкозурией, выраженной дегидратацией организма и задержкой роста. Необходимость инсулинотерапии исчезает в течение 6 недель. Выздоровление происходит у 50% новорожденных к 1,5 годам, но у пациентов остается предрасположенность к развитию сахарного диабета 2-го типа в течение взрослой жизни.
ТНСД имеет три типа, которые обусловлены патологией в разных генах. ТНСД1 является результатом аномальной экспрессии импринтированного гена PLAGL1 в районе хромосомы 6q24.2 или гена ZFP57 в районе хромосомы 6p22.1. ТНСД типа 2 и 3 возникает в результате мутаций в генах ABCC8 и KCNJ11, соответственно, расположенных в районе хромосомы 11р15.1 и не относятся к болезням импринтинга. ТНСД1 возникает в результате отцовских ОРД хромосомы 6 (35-40%), дупликации 6q24 (35–40%) и аномалий метилирования ЦИ материнского аллеля в 6q24 (20%). Мутации ZFP57 достаточно редки и описаны всего в нескольких случаях ТНСД.
Несколько спорадических случаев ТНСД с отцовской ОРД по хромосоме 6 позволили предположить, что заболевание связано либо с экспрессией двойной дозы отцовских импринтированных генов, либо с отсутствие экспрессии таковых на материнской хромосоме (рис. 20).
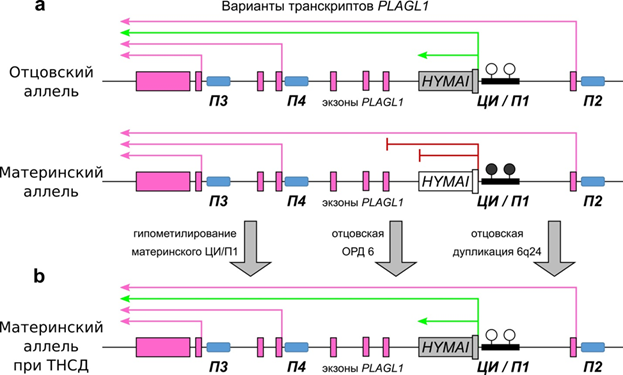
Рисунок 20. Структура импринтированного района 6q24.2 и варианты экспрессии генов при ТНСД (пояснения в тексте). Обозначения: розовые прямоугольники и стрелки – биаллельно экспрессирующиеся последовательности; зеленые стрелки – отцовская экспрессия; черные и белые кружки – метилированное и неметилированное состояние ЦИ; П1 – П4 промоторные районы.
В небольшом числе описанных семейных случаев наследование было исключительно от отца и ассоциировалось с дупликацией района хромосомы 6q24. Молекулярный анализ таких семей позволил определить наименьший район дупликаций, составляющий 500 тыс. пар нуклеотидов. В нем был обнаружен ДМР, метилированный на материнской хромосоме, и неметилированный на отцовской хромосоме, регулирующий два импринтированных гена - PLAGL1 и HYMAI, которые активно экспрессируются в поджелудочной железе.
Ген PLAGL1 регулирует транскрипцию и является геном-супрессором опухолевого роста. Его белок с мотивом цинковых пальцев индуцирует транскрипцию гена PACAP (рецептор полипептида, активирующего аденилатциклазу гипофиза), который усиливает секрецию инсулина и регулирует работу β-клеток поджелудочной железы. Гиперэкспрессия PLAGL1 приводит к аномальной работе этих клеток. Кроме того, PLAGL1 регулирует работу ряда импринтированных и не импринтированных генов, таких, как IGF2, H19, SLC2A4, CDKN1C и PPARγ1, вовлеченных в процессы клеточного роста и метаболизма. PLAGL1 состоит из 12 экзонов и экспрессируется с четырех различных промоторов, в результате чего получаются 4 изоформы белка. Транскрипты с промоторов П2, П3 и П4 экспрессируются биаллельно и только транскрипт с П1, который является ДМР и ЦИ, экспрессируется только с отцовского аллеля. Еще один ген в этом локусе – HYMAI кодирует днРНК. Ген состоит из одного экзона и имеет перекрывающийся с транскриптом PLAGL1 сайт старта транскрипции в ЦИ/П1. Поэтому статус метилирования ЦИ/П1 регулирует, как экспрессию HYMAI, так и определенных транскриптов PLAGL1.
Обычно отцовский ЦИ/П1 гипометилирован, а материнский гиперметилирован, следовательно, транскрипты HYMAI и PLAGL1, регулируемые ЦИ/П1, экспрессируются с отцовского аллеля. Потеря метилирования материнского аллеля у пациентов ТНСД сопровождается повышенным весом и индексом массы тела.
МНОЖЕСТВЕННЫЕ АНОМАЛИИ МЕТИЛИРОВАНИЯ ИМПРИНТИРОВАННЫХ РЕГУЛЯТОРНЫХ РАЙОНОВ.
С 2006 г. начали обсуждать вопрос о новой болезни импринтинга, являющейся результатом материнского гипометилирования различных импринтированных локусов - множественные аномалии метилирования (МАМ). МАМ были выявлены в импринтированных, как материнских, так и отцовских, локусах при различных болезнях импринтинга. Наибольшая частота эпимутаций наблюдается при ТНСД (50%), гипометилирование ЦИ2 при СВБ (25%). МАМ при СРС и ПГПТ1В определяется в 8-10%. Была описана семья (близкородственный брак), в которой две дочери имели фенотипические признаки ТНСД с некоторыми симптомами СБВ. При исследовании статуса метилирования импринтированных районов установлено, что потеря метилирования произошла не только в импринтированном районе ZAC (6q24), но и в районах KCNQ1OT1 (11p15.5), GRB10 (7p11.2-р12), PEG3 (19q13), PEG1/MEST (7q32) и NESPAS (20q13). Было предположено, что в семье существовал некий аутосомнорецессивный дефект, повреждающий механизмы метилирования у потомства, или был нарушен процесс установления импринтинга в ооцитах.
В результате исследований были описаны мутации в гене ZFP57 у пациентов с ТНСД и МАМ в разных ЦИ. Ген ZFP57 кодирует транскрипционный репрессор, который формирует комплекс с белком ко-репрессором KAP1 (KRAB-ассоциированный белок-1). KAP1 рекрутирует другие белки, такие, как метилтрансфераза лизина 9 гистона Н3 (SETDB1), ядерный белок NP95, который, в свою очередь, привлекает ДНК-метилтрансферазы. Таким образом, этот белковый комплекс играет огромную роль в регуляции и поддержания метилирования ДНК в различных ЦИ. Поэтому мутации ZFP57, приводящие к потере белка или появлению дефектного белка, нарушают метилирование различных ЦИ, что приводит к потере импринтинга.
ZFP57 связывается с метилированными ЦИ в предимплантационном развитии, защищая их от деметилирования и сохраняя родительскую идентичность. В норме сайт для связывания ZFP57 обнаружен в районах 17 из 31 импринтированных ДМР, а в результате его мутаций наиболее часто нарушается метилирование PEG3, PLAGL1, INPPF5, NAP1L5 и GRB10.
Профиль экспрессии еще одного члена семейства белков с мотивом цинкового пальца, ZF P 445, устойчивость к мутациям с потерей функции, возможность белка связываться и обеспечивать (формировать) гетерохроматин в районах ЦИ говорит о его важной роли в поддержании метилирования на ранних этапах развития эмбриона. Нокдаун гена приводит к невозможности связывания KAP1 и метилирования H3K9me3, следовательно, увеличивается экспрессия генов, происходит потеря метилирования в ЦИ, включая Н19. Все это подтверждает, что, подобно ZFP57, ZFP445 может связываться с ЦИ, привлекать KAP1 и запускать метилирование H3K9me3.
Также показано, что нокаут ZFP42 вызывает потерю метилирования нескольких импринтированных ДМР, за исключением Н19, а единственная выявленная мутация привела к МАМ.
Еще одной возможной причиной множественных аномалий метилирования ЦИ могут являться белки NLRP, представляющие собой небольшое подсемейство белков, необходимых для созревания ооцита и регулирования метилирования на ранних стадиях эмбриогенеза. Материнская мутация NLRP2 была обнаружена у двух детей с СБВ и МАМ. В результате мутаций NLRP5, и материнские, и отцовские импринтированные ДМР теряют метилирование, что так же приводит к МАМ. NLRP7 вовлечен в установление ооцит-специфического метилирования, а его мутации приводят к рецидивирующему пузырному заносу с широкой потерей материнского метилированного импринта, в то время как отцовские метилированные ДМР не изменяются.
РЕДКИЕ СИНДРОМЫ, СВЯЗАННЫЕ С ИМПРИНТИРОВАННЫМИ ГЕНАМИ.
Синдром Бирк-Барел (СББ) (OMIM #612292) характеризуется тяжелой гипотонией новорожденных, преходящей гипогликемией новорожденных, контрактурами суставов, широкими альвеолярными гребнями, расщелиной неба, микроретрогнатией, задержкой развития и вариабельной умственной отсталостью. Отличительными чертами лица являются долихоцефалия, битемпоральное сужение, короткий фильтр, нависающая верхняя губа и медиально выступающие брови. Это заболевание вызвано специфической миссенс-мутацией (c.770G>A, p.Gly236Arg) в материнской копии гена KCNK9/TASK3, расположенного в хромосомном районе 8q24. В этом районе имеется два импринтированных гена: PEG13 экспрессируется с отцовского аллеля, а KCNK9 - с материнского аллеля. Реципрокная экспрессия этих генов регулируется материнским ДМР, расположенным внутри транскрипта PEG13.
Ген KCNK9/TASK3 кодирует белок подсемейства калиевых каналов с двумя поровыми доменами. Каналы TASK3 широко экспрессируются, особенно в головном мозге, где они играют роль в миграции кортикальных пирамидальных нейронов. Следует отметить, что нестероидные противовоспалительные препараты фенаминовой кислоты, особенно тефлуфенаминовая кислота, способны стимулировать двух-поровые калиевые каналы, частично восстанавливая уменьшенный внешний ток через KCNK9-мутантные каналы, а это свидетельствует о том, что соединения фенаминовой кислоты могут быть полезны при лечении этого состояния.
Синдром задержки внутриутробного развития, метафизарной дисплазии, врожденной гипоплазия надпочечников и аномалий половых органов - IMAGE (OMIM #614732) является результатом миссенс-мутаций в гене CDKN1C, который отрицательно регулирует клеточную пролиферацию. Все 5 мутаций кластеризованы в высоко консервативной области в пределах 6 аминокислот PCNA-связывающего домена CDKN1C и нарушает связывание PCNA (ядерный антиген пролиферирующих клеток). Поскольку CDKN1C, расположенный в критическом районе для СБВ (хромосома 11р15.4) экспрессируется только с материнского аллеля, синдром возникает только тогда, когда мутация наследуется от матери. Синдром характеризуется СРС-подобным фенотипом, метафизарной дисплазией, врожденной гипоплазией надпочечников с надпочечниковой недостаточностью, при этом часто выявляются половые аномалии (двусторонний крипторхизм, микропенис и гипогонадотропный гипогонадизм.
Ретинобластома (РБ) (#ОMIM 180200) – наиболее частая детская интраокулярная злокачественная опухоль сетчатки глаза, составляющая 3% всех опухолей детского возраста. Опухоль возникает из клеток-предшественников колбочек с частотой 1:16000-18000 новорожденных и характеризуется высокой степенью злокачественности, инвазивностью и способностью быстро метастазировать в соседние органы и ткани.
В основе молекулярных событий, приводящих к развитию РБ, лежит биаллельная инактивация гена-супрессора RB1 в клетках опухоли. При спорадической РБ, которая составляет 60% случаев, обе мутации являются соматическими, то есть происходят в одной и той же клетке-предшественнице сетчатки. Спорадическая форма проявляется как унилатеральная опухоль и диагностируется, как правило, после 2 лет.
При наследственной РБ, обнаруживаемой в 40% случаев, герминальная мутация в одном из аллелей гена RB1 обуславливает предрасположенность к заболеванию и его семейную передачу, а развитие опухоли инициируется соматической мутацией в другом аллеле гена в клетке сетчатки, которая приобретается внутриутробно или в раннем детстве. Наследственная РБ манифестирует в более раннем возрасте по сравнению со спорадической формой (средний возраст постановки диагноза составляет 12 месяцев) и носит в большинстве случаев мультифокальный и билатеральный характер. Заболевание передается аутосомно-доминантно с пенетрантностью более 90%. Кроме того, у носителей герминальной мутации повышен риск развития других первичных опухолей различной локализации.
В некоторых семьях с РБ (два и более носителя одинаковой герминальной мутации в родословной) возможен более мягкий фенотип с неполной пенетрантностью (у части носителей герминальной мутации заболевание не развивается) и вариабельной экспрессивностью (одинаковая мутация у разных членов семьи может проявляться уни- или билатеральной формой заболевания). Фенотипическое проявление наследственной РБ зависит от функционального типа герминальной мутации в гене RB1. В свою очередь, молекулярные механизмы, лежащие в основе вариабельного фенотипического проявления одинаковой мутации у разных членов семьи объясняются эффектом родительского происхождения мутации RB1.
В гене RB1 идентифицирован импринтированный регион (1,2 т.п.н.) - CpG-островок (CpG 85), расположенный во втором интроне гена, который дифференциально метилирован в зависимости от родительского происхождения аллеля - регион метилирован на материнской хромосоме и неметилирован на отцовской. Кроме того, RB1 содержит еще два CpG-островка: CpG 106 и CpG 42. Островок CpG 106, захватывающий промотор и 1-й экзон гена, биаллельно неметилирован и обеспечивает биаллельную экспрессию основного транскрипта, кодирующего белок pRB. Островок CpG 42, располагающийся во 2-м интроне гена, метилирован на обеих хромосомах и не обладает регуляторной активностью (рис. 21).
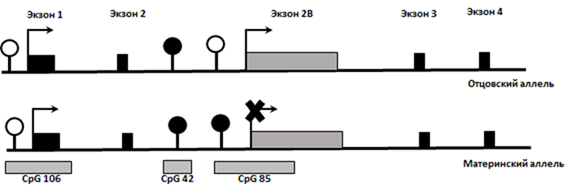
Рисунок 21. Структура 5’- района гена RB1 и локализация в нем CpG-островков (CpG 106, CpG 42, CpG 85). Экзоны 1-4 - регулярные экзоны RB1, экзон 2B - стартовый экзон для альтернативного транскрипта гена (первый экзон альтернативного транскрипта 2B соединяется с 3 регулярным экзоном RB1). Белыми кружками обозначены неметелированные CpG-островки, черными - метилированные; стрелки указывают направление транскрипции.
Установлено, что CpG 85 является частью 5’-усеченного процессированного псевдогена, образовавшегося из белок-кодирующего гена KIAA0649, расположенного на 9 хромосоме, и интегрировавшего в RB1 в обратной ориентации. CpG 85 выступает в качестве промотора для альтернативного транскрипта RB1, который экспрессируется только с неметилированной отцовской хромосомы. Несмотря на ожидаемо более высокий общий уровень экспрессии мРНК транскриптов с отцовского аллея по сравнению с таковым с материнского, уровень экспрессии с отцовского аллея ниже в 2-3 раза за счет интерференции транскрипции, вызванной совместной экспрессией регулярного и альтернативного транскриптов. Интерференция транскрипции – это механизм, при котором транскрипция одного гена имеет подавляющее влияние на транскрипцию другого гена. Так, транскрипционный комплекс альтернативного транскрипта гена RB1, который связывается с неметилированным островком CpG 85 на отцовской хромосоме, выступает в качестве блока для прохождения транскрипционного комплекса основного транскрипта на том же аллеле, что приводит к уменьшению уровня экспрессии на отцовской хромосоме. Это свидетельствует об эпигенетической регуляции экспрессии гена RB1 в зависимости от родительского происхождения аллеля.
В частности, низкопенетрантные герминальные мутации RB1, унаследованные от отца, приводят к развитию РБ чаще и в более тяжелой форме по сравнению с таковыми, унаследованными от матери. Считается, что в случае наследования низкопенетрантной мутации по материнской линии, опухолевой супрессорной активности pRB достаточно для предотвращения развития РБ. Напротив, при передаче мутантного аллеля от отца, низкая остаточная активность белка за счет более низкой экспрессии будет имитировать мутацию, приводящую к отсутствию белка, что приведет к развитию заболевания.
Синдром Шаафа-Янга (СШЯ) (OMIM #615547) является СПВ-подобным заболеванием, обусловленным нонсенс- и миссенс-мутациями (всего 9) в гене MAGEL2, который расположен в критической области хромосомы 15q11–q13 и обычно импринтирован на материнской хромосоме, а экспрессируется с отцовской. Синдром характеризуется гипотонией новорожденных, задержкой развития и умственной отсталостью, гипогонадизмом, аутистическим поведением и контрактурами суставов. Типичные проявления СПВ, такие, как гиперфагия и ожирение обычно отсутствуют; таким образом фенотипическое сходство с СПВ ограничивается неонатальным периодом. Фенотипические проявления варьируют от тяжелой фетальной акинезии до легкой степени выраженности, включая умственную отсталость и контрактуры пальцев. Интересно, что мутации, приводящие к укороченному белковому продукту гена MAGEL2 вызывают СШЯ, а делеции всего гена приводят лишь к незначительным проявлениям или полному отсутствию клинического фенотипа. Поскольку MAGEL2 имеет только один экзон, мутации могут приводить к укороченному белку с доминантно-негативным эффектом. Так же, вполне вероятно, что делеция всей отцовской копии гена, включая его промотор, может привести к частичному восстановлению экспрессии материнского метилированного аллеля.
Синдром центрального преждевременного полового созревания (CPP) (OMIM #176400), также известный, как гонадотропин зависимое преждевременное половое созревание, характеризуется преждевременной активацией репродуктивной оси, до 6-летнего возраста (5-6,5 лет) у девочек и 8-летнего (5,9-8,5) - у мальчиков. Распространенность CPP оценивалась примерно в 1,1 на 100 000, при общем соотношении мужчин и женщин, как 1:10. У пациентов выявляют признаки раннего полового созревания, такие как преждевременное развитие молочных желез или увеличение яичек, ускорение роста и костного возраста, сопоставимые с повышенными базальным и GnRH-стимулированным уровнями ЛГ. Центральное преждевременное половое созревание 2 (CPPB2) (OMIM #615346) обычно вызывается гетерозиготными инактивирующими мутациями в гене MKRN3/ZFP127, расположенном в критической области СПВ (хромосома 15q11–q13). Антисмысловая РНК ZNF 127 AS с неизвестной функцией перекрывает этот ген и, вероятно, регулирует экспрессию MKRN 3/ZFP 127. Ген импринтирован на материнской хромосоме, а экспрессируется с отцовской, поэтому только мутации, унаследованные от отцов, приводят к заболеванию. Только один выявленный носитель мутации, унаследованной от матери, не имел фенотипических проявлений. Высокая частота мутаций MKRN3/ZFP127 была обнаружена в когорте мужчин с преждевременным половым созреванием. Половое созревание обычно начинается с импульсного выхода GnRH из гипоталамических нейронов. Действительно, наступление половой зрелости требует, как уменьшения факторов, которые ингибируют высвобождение GnRH, так и увеличения стимулирующих факторов. Уровни MKRN3/ZFP127 снижались до клинического начала, в период полового созревания и отрицательно коррелировали с гонадотропинами у препубертатных девочек; их уровни в крови снижались во время полового созревания у здоровых мальчиков. Характер экспрессии MKRN3/ZFP127 оказывает ингибирующее влияние на секрецию GnRH, но точный механизм, посредством которого его дефицит приводит к ранней реактивации пульсирующей секреции GnRH, еще предстоит выяснить. Агонисты GnRH могут быть вариантом лечения CPP для того, чтобы уменьшить скорость костного возраста, роста и прогрессирования клинических признаков полового созревания.
Дата: 2019-12-10, просмотров: 671.