1. Наиболее распространенная причина возникновения СПВ и СА - протяженная делеция критического района 15(q11.2-q13). Ее обнаруживают у 60-75% пациентов, а в популяции - с частотой 0,67-1,0 на 10 000 новорожденных. Это позволяет считать ее одной из самых частых хромосомных перестроек у человека. Причиной СПВ становится делеция критического района на отцовской хромосоме 15, а СА возникает в случае делеции той же области на ее материнском гомологе.
Спорадический характер возникновения делеции критического района хромосомы 15(q11.2-q13) при СПВ и СА, ее стандартная протяженность в 90% случаев, строгая локализация точек разрыва и одинаковая частота выявления при СПВ и СА указывают на то, что возникновение делеции происходит в гаметогенезе родителей (одинаково часто в овогенезе и сперматогенезе) и объясняется мейотической нестабильностью этого хромосомного района.
Делеции являются результатом негомологичной рекомбинации, обусловленной блоками повторяющихся последовательностей, расположенных в районах точек разрыва (BP1–BP5). ВР1 и ВР2 локализуются проксимальнее MKRN3, а ВР3, 4 и 5 – теломерней. Выделяют 2 класса делеций: делеции 1 класса составляют 40% и происходят в ВР1-ВР3; делеции 2 класса составляют 50% и находятся между ВР2 и ВР3. В редких случаях (менее 10%) делеции достигают 10,6 млн.п.н. и простираются теломерней ВР3 вплоть до ВР4 и ВР5. Дистальный кластер точек разрыва локализован теломернее гена OCA2, отвечающего за пигментацию организма. Такие делеции включают кластер из трех генов-рецепторов ГАМК - GABRB3, GABRA5 и GABRG3. Наиболее частые точки разрыва BP1, BP2 и BP3 фланкированы низкокопийными повторами, произошедшими из дуплицированных фрагментов гена HERC2, первоначальная копия которого располагается в ВР3. ВР4 и ВР5 тоже содержат низкокопийные повторы, но не имеющие гомологии с HERC2. Делеции являются результатом внутри- или межхромосомного кроссинговера, происходят de novo, благодаря образованию большой (4-5 млн пар нуклеотидов) петли и только в редких случаях – результатом структурной перестройки.
Кроме того, описано большое количество случаев с вовлечением района 15(q11.2-q13), не только в делеции, но и в инвертированные дупликации (тетрасомия), дупликации (трисомия), несбалансированные (моносомия) и сбалансированные транслокации, а также в инверсии.
2. Вторая причина развития СПВ и СА - ОРД, т.е. наследование обоих гомологов хромосомы 15 от одного из родителей. Отцовская ОРД становится причиной заболевания у 3-5% пациентов с СА, а материнская ОРД - в 25% случаев СПВ. В силу функциональной неравнозначности отцовской и материнской хромосомы 15 существование у ребенка двух структурно полноценных хромосом материнского происхождения не обеспечивает нормального развития и, как в случае отцовской делеции, приводит к развитию СПВ. Аналогично этому отцовская ОРД приводит к развитию СА. У большинства больных с СПВ, имеющих ОРД, регистрируют гетеродисомию, возникающую вследствие нерасхождения материнских хромосом в первом делении мейоза. Отцовская ОРД при СА, как правило, манифестирует изодисомией и возникает путем коррекции моносомии до дисомии. Так как зиготы, несущие моно- и трисомию по хромосоме 15, нежизнеспособны, удвоение единственной хромосомы при моносомии и потерю лишней хромосомы при трисомии можно считать «аварийными» мерами, после которых становится возможным дальнейшее развитие эмбриона.
Несмотря на то, что и материнская ОРД при СПВ, и отцовская ОРД при СА возникают вследствие нерасхождения хромосом в гаметогенезе, ОРД у пациентов с СПВ регистрируют значительно чаще. Это можно объяснить большей выживаемостью зигот с трисомией по хромосоме 15 и ранней гибелью моносомных зигот. Кроме того, потеря лишней хромосомы в трисомной клетке, по-видимому, более вероятное событие, чем удвоение хромосомы в моносомной клетке на ранней стадии развития.
3. Были описаны пациенты (менее 5%) с клиническим диагнозом СА и СПВ, у которых не было типичных делеций района 15(q11.2-q13) и ОРД, но, как и в случае ОРД, присутствовал одинаковый характер метилирования обеих родительских копий этой критической области. Это объясняли нарушением переключения (заменой) эпигенотипа хромосом в гаметогенезе одного из родителей вследствие ошибок импринтинга - эпимутации. Если в сперматогенезе отца на его материнской хромосоме не происходит замены женского импринта на мужской, то в следующем поколении, после объединения с материнским геномом, возникнет состояние, аналогичное материнской ОРД и функционально равнозначное отцовской делеции, которое будет сопровождаться фенотипом СПВ. Нарушение установления женского эпигенотипа на отцовских хромосомах в овогенезе матери приведет к развитию СА у потомства.
Молекулярно-генетические исследования у таких пациентов показали, что ошибки импринтинга сопровождаются микроделециями особой регуляторной области, расположенной в пределах СА/СПВ-кластера импринтированных генов - ЦИ.
В небольшом количестве случаев аномалия импринтинга вызвана микроделециями, нарушающими ЦИ, который регулирует установление и поддержание импринта во всем критическом районе 15q11.2–q13. Наименьшая область перекрывания делеций ЦИ, достаточная для возникновения СА, соответствует ЦИ-СА. Делеция этого района не позволяет установить материнский импринт на поврежденной хромосоме. Было обнаружено, что ЦИ-СА служит ооцит-специфическим промотором для некодирующего транскрипта, который перекрывает район промотора/экзона 1 гена SNRPN. В растущем ооците, сквозная транскрипция требует присутствия метилтрансферазы DNMT3A и кофактора DNMT3L для метилирования этого района. Выявлено всего несколько делеций, которые произошли de novo на материнской хромосоме, или были результатом мозаичной делеции, произошедшей в герминальных клетках у матери. Более 94% дефектов импринтинга, приводящих к СА, относятся к первичной эпимутации без каких либо изменений на уровне ДНК. В этих случаях риск рецидива в семьях очень низок.
Собственно делеции ЦИ не сопровождаются развитием патологического фенотипа и могут существовать у здоровых родителей и других родственников пациента. Анализ родословных тех семей с СПВ и СА, где есть делеции ЦИ, свидетельствует о том, что они могут наследоваться в семьях без фенотипических признаков через несколько поколений одного и того же пола (рис. 12). Например, делеция ЦИ-СА - области, ответственной за установление материнского эпигенотипа, - может наследоваться через несколько поколений (от отца к сыну) до тех пор, пока не будет передана дочери. В этом случае дочь будет здорова, но в ее овогенезе импринт отцовской хромосомы не заменится на материнский. Ребенок этой женщины (независимо от пола) с вероятностью 50% получит от матери хромосому с ошибочным отцовским эпигенотипом и будет иметь клиническую картину СА. Аналогично этому возможно скрытое наследование делеций ЦИ-СПВ в ряду поколений от матери к дочери, но риск рождения больного ребенка с СПВ у мужчины-носителя такого нарушения также составляет 50%.
Все случаи СПВ и СА вследствие ошибок импринтинга, но без делеций ЦИ, спорадические. Действительно, есть сообщения о том, что здоровые сибсы могут наследовать ту же хромосому, которая у больного брата или сестры несет ошибочный эпигенотип. Таким образом, риск повторного рождения больного ребенка в таких семьях
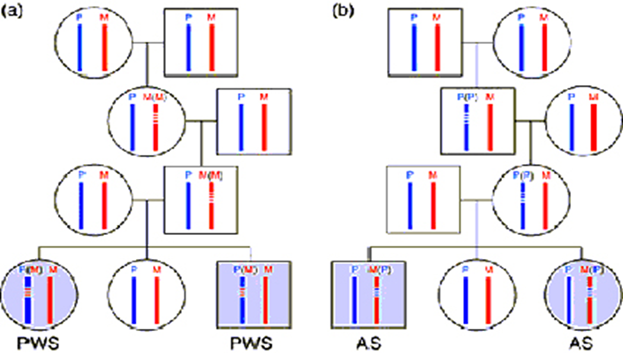
Рисунок 12. Наследование мутаций центра импринтинга, приводящих к невозможности переключения импринта в герминальных клетках: а — при синдроме Прадера–Вилли, если мутация возникает во втором поколении у женщины, то она фиксирует материнский импринт М (М), который передается без фенотипических последствий следующему поколению; у мужчины мутация блокирует стирание женского импринта, поэтому 50% его потомков будут страдать этим синдромом и иметь эпигенотип Р (М); б — при синдроме Ангельмана отцовский импринт Р (Р) фиксируется и может передаваться без аномалий фенотипа через мужчин, но в герминальных клетках женщины мутация не позволяет изменить эпигенотип М (Р), что в 50% случаев приводит к рождению больного ребенка. PWS — синдром Прадера–Вилли; AS — синдром Ангельмана; P — отцовская хромосома; M — материнская хромосома.
крайне мал, а в случае делеции ЦИ он равен 50%. Целесообразность выделения двух групп пациентов на основе существования или отсутствия делеций ЦИ объясняют необходимостью расчета риска повторного рождения больных детей, от которого зависит тактика генетического консультирования семьи. Высказывают предположение о том, что спорадические случаи ошибок импринтинга без делеций ЦИ - результат случайных сбоев в многоэтапном процессе становления эпигенотипа, которые могут быть связаны со структурными нарушениями в других (помимо ЦИ) цис-регуляторных последовательностях или дефицитом пока неизвестных внехромосомных транс-действующих факторов.
4. Мутации в гене UBE3A установлены в 5-10% случаев СА и представляют собой мутации материнского аллеля с преждевременным терминированием синтеза белка. Большинство выявленных мутаций нарушают домен HECT- лигазы. Около 29% мутаций наследуются от матери и 71% возникают de novo. Атипичные делеции UBE3A или редкие типы структурных перестроек, нарушающих критический район, были зарегистрированы только у нескольких пациентов с семейным СА. Как было сказано выше, точковых мутаций в генах импринтированного района, которые бы соответствовали фенотипу СПВ, не выявлено.
Дата: 2019-12-10, просмотров: 366.