Понятие о коэффициенте калорийности пищи. Коэффициенты калорийности основных компонентов пищи: БЖУ.
Коэффициент калорийности – энтальпия сгорания 1 г вещества, взятая с обратным знаком. Выражается в кДж/г или ккал/г (1 ккал = 4,18 Дж).
Коэффициенты калорийности:
- белков 16,5 – 17,2 кДж/г;
- углеводов 16,5 – 17,2 кДж/г;
- жиров 37,7 – 39,8 кДж/г.
Для расчета калорийности порции пищи, используют формулу:
Кmin = (mб · 16,5 + mу · 16,5 + mж · 37,7) кДж
Кmax = (mб · 17,2 + mу · 17,2 + mж · 39,8) кДж
Второй закон термодинамики, его формулировки. Энтропия и энергия Гиббса как критерии возможности самопроизвольного протекания процессов.
Формулировки:
1. Теплота не может сама собой передаваться от холодного тела к горячему, не оставляя изменений в окружающей среде.
2. Различные виды энергии стремятся перейти в теплоту, а теплота рассеивается, т.е. её невозможно полностью превратить в полезную работу.
3. В изобарно-изотермических условиях самопроизвольно протекают только такие процессы, которые сопровождаются уменьшением энергии Гиббса (G<0). В состоянии равновесия энергия Гиббса не меняется, ΔG=0.
Энтропия ( S) – мера неупорядоченности в системе. Это функция состояния системы, приращение которой (ΔS) равно минимальной теплоте Q, поступившей в систему в обратимом изотермическом процессе, делённой на абсолютную температуру (T), при которой совершается этот процесс. ΔS=Qmin/T, [Дж · моль-1 · К-1].
Энергия Гиббса ( G) - это часть потенциальной энергии реагирующих веществ, которая может быть использована для осуществления полезной работы.
В изобарно-изотермических условиях ΔG = ΔH – TΔS.
Самопроизвольные процессы протекают без сообщения энергии системе извне. Протекают до установления равновесия.
Величина ΔG служит критерием возможности самопроизвольного протекания процессов.
Если ΔG<0 – процесс протекает самопроизвольно.
Если ΔG>0 – процесс самопроизвольно не протекает.
Если ΔG=0 – состояние равновесия.
Химическое равновесие, константа равновесия. Термодинамическая характеристика химического равновесия. Уравнение изотермы химической реакции, условия равновесия и направления обратимых химических реакций.
Химическое равновесие – состояние системы, когда пряма и обратная реакции имеют одинаковые скорости.
Состояние химического равновесия характеризуется константой равновесия химической реакции, которая равна отношению констант скоростей прямой и обратной реакций: Kр = kпрям / kобр.
Константа равновесия – постоянная, отражающая соотношение концентраций компонентов обратимой реакции в состоянии химического равновесия.
Кс = [Сс][Dd]/[Aa][Bb]
Уравнение изотермы: 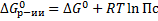
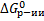 – изменение Е Гиббса
– изменение Е Гиббса
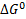 – изменение стандартной Е Гиббса
– изменение стандартной Е Гиббса
Пс – величина стехиометрического соотношения концентраций веществ, участвующих в реакции
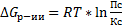
Анализ:
1) Если Пс>Кс, Пс/Кс > 1, lnПс/Кс > 0, то > 0 – обратная реакция.
2) Если Пс<Кс, Пс/Кс < 1, lnПс/Кс < 0, то < 0 – прямая реакция.
3) Если Пс=Кс, Пс/Кс = 1, lnПс/Кс = 0, то = 0 – равновесие в системе.
Константа равновесия реакции связана со свободной энергией Гиббса: ΔG = – RT · ln Kр. Если Kр = 1, то ΔG = 0, и протекание реакции равновероятно в обе стороны. Если Kр > 1, то ΔG < 0, и реакция смещена в сторону образования продуктов. Если Kр < 1, то ΔG > 0, реакция протекает преимущественно в сторону образования реагентов, то есть преобладает обратная реакция.
Минимальное значение энергии Гиббса является условием термодинамического равновесия в закрытой системе.
Если Кс = 0, реакция не идёт.
Если Кс = ∞, реакция идёт до конца.
Если Кс > 0, реакция прямая, равновесие смещено в образование продуктов.
Если Кс < 0, реакция обратная, равновесие смещено в образование реагентов.
Электролиты. Степень диссоциации. Константа диссоциации. Сильные электролиты. Состояние ионов в растворах сильных электролитов. Межионное взаимодействие. Понятие об активности, коэффициент активности.
Электролиты – это вещества, водные растворы или расплавы которых проводят электрический ток (кислоты, основания, соли).
Электролитическая диссоциация – распад молекул на ионы.
Степень диссоциации электролита ( α ) - показывает отношение числа молекул электролита, распавшихся на ионы (n), к общему числу диссоциированных (n) и недиссоциированных (N) молекул:
Величина α зависит от природы электролита, температуры и концентрации вещества в растворе.
Классификация электролитов по величине степени диссоциации (α):
1. Сильные электролиты: α > 0,3 (30%), в разбавленных растворах α -> 1 (100%) – сильные кислоты, растворимые основания – щёлочи, все растворимые соли.
2. Электролиты средней силы: 0,3 > α > 0,03 (от 3 до 30 %)
3. Слабые электролиты: α < 0,03 (<3%)
Константа диссоциации: Чем больше величина Кд, тем сильнее диссоциирует электролит.
На практике используют величину рКд (показатель константы диссоциации):
Чем выше значение рКд, тем слабее диссоциирует электролит.
Кд зависит от природы электролита, температуры и практически не зависит от концентрации вещества в растворе.
Для слабых электролитов справедлив закон Оствальда: степень диссоциации слабого электролита возрастает с разбавлением раствора.
Активность – это концентрация ионов в растворе с учётом сил межионного взаимодействия.
a(x) = f(x) * C(x), где a(x) – активность электролита, моль*дм(-3), С(х) – концентрация электролита, моль*дм(-3), f(x) – коэффициент активности - выражает отклонение свойств раствора с концентрацией С(х) от свойств идеального бесконечно разбавленного раствора данного электролита. Принимает значения от 0 до 1.
Если f(х)=1, тогда а(х)=С(х), ионы практически не связаны межионным взаимодействием. Это достигается в разбавленных растворах (C(x)≈10-4 моль·дм-3).
Если f(x)<1, тогда a(x)<C(x), ионы связаны силами межионного взаимодействия. При этом во всех расчетах используют именно активную концентрацию, меньшую по числовому значению, чем C(x).
Растворы, применяемые в медицинской практике и имеющие ионную силу равную 0,15 моль·кг-1, называются физиологическими растворами. I физ = 0,15 моль·кг-1.
Буферные системы, их состав, свойства, классификация. Механизм буферного действия. Буферные растворы. Уравнение кислых буферных систем Гендерсона-Гассельбаха, его вывод и анализ. Зона буферного действия. Буферная ёмкость. Факторы, влияющие на величину буферной ёмкости.
Буферные системы – это системы, состоящие из 2х сопряжённых компонентов, способных до определённого предела противодействовать изменению рН среды при добавлении к ним небольших количеств кислоты и щелочи, а также при разбавлении раствора или концентрировании.
Классификация:
- по составу:
1)Кислые – состоят из слабой кислоты и сопряжённого с ней избытка сильного основания, создаваемого солью этой кислоты. Например:
Ацетатная буферная система:
СН3СООН – слабая кислота;
СН3СООNa – растворимая соль (содержит сопряженное сильное основание СН3СОО- ).
Карбонатная буферная система:
Н2СО3
NaНСО3
Фосфатная буферная система:
NaН2РО4
Na2НРО4
Белковая буферная система:
Белок-Н или Prot-Н
Белок-Na Prot-Na
2) Основные – состоят из слабого основания и сопряжённого с ним избытка кислоты, создаваемого солью этого основания. Например:
Аммиачная буферная система:
NН4ОН – слабое основание;
NН4Сl – растворимая соль (содержит сопряженную сильную кислоту NН4+).
Буферные растворы – это буферные системы в растворённом состоянии. В отличие от системы, растворы могут быть многокомпонентными, например, кровь.
Буферное действие – способность буферных систем сохранять постоянство рН.
Механизм буферного действия: постоянство pH поддерживается за счет того, что избыток свободных ионов H+ или OH- связывается одним из компонентов буферной системы в малодиссоциирующее соединение.
Классификация КС по различным признакам. Заряд комплексного иона. Катионные, анионные, нейтральные КС, их номенклатура. Моно- и полидентантные лиганды. Хелаты. Комплексоны. Трилон Б. Краун-эфиры. Значение комплексных соединений в биологии и медицине.
Классификация:
1. По составу: соли, основания, кислоты.
2. По типу координируемых лигандов:
а) Аквакомплексы – это комплексные катионы, в которых лигандами являются молекулы H2O. Их образуют катионы металлов со степенью окисления +2 и больше, причем способность к образованию аквакомплексов у металлов одной группы периодической системы уменьшается сверху вниз.
Например: [Al(H2O)6]Cl3, [Cr(H2O)6](NO3)3.
б) Гидроксокомплексы – это комплексные анионы, в которых лигандами являются гидроксид-ионы OH–. Комплексообразователями являются металлы, склонные к проявлению амфотерных свойств – Be, Zn, Al, Cr.
Например: Na[Al(OH)4], Ba[Zn(OH)4].
в) Аммиакаты – это комплексные катионы, в которых лигандами являются молекулы NH3. Комплексообразователями являются d-элементы.
Например: [Cu(NH3)4]SO4, [Ag(NH3)2]Cl.
г) Ацидокомплексы – это комплексные анионы, в которых лигандами являются анионы неорганических и органических кислот.
Например: K3[Al(C2O4)3], Na2[Zn(CN)4], K4[Fe(CN)6].
3. По характеру заряда внутренней сферы различают катионные, анионные и нейтральные комплексы.
Например:
1) [Cu+( NН3)4]+ - катионный комплекс
2) [Zn2+(SCN)4]2– - анионный комплекс
3) [ Pt2+(CI)2(Н 2 О)2]0 - нейтральный комплекс
Номенклатура катионных комплексов:
1. Назвать число лигандов греческим числительным
2. Назвать лиганды: сначала нейтральные молекулы, затем лиганды-анионы
3. Назвать комплексообразователь русским наименованием
4. Отметить валентность комплексообразователя римской цифрой в скобках
Номенклатура анионных комплексов:
1. Назвать число лигандов греческим числительным
2. Назвать лиганды: сначала нейтральные молекулы, затем лиганды-анионы
3. Назвать комплексообразователь латинским наименованием с окончанием -ат
4. Отметить валентность комплексообразователя римской цифрой в скобках
Номенклатура нейтральных комплексов:
1. Назвать число лигандов греческим числительным
2. Назвать лиганды: сначала нейтральные молекулы, затем лиганды-анионы
3. Назвать комплексообразователь русским наименованием
Монодентантные лиганды присоединяются к комплексообразователю одним атомом и образуют одну координационную связь. (Например: H2O, NH3, Cl-, CN-, OH- и тд.)
Полидентантные присоединяются к комплексообразователю посредством нескольких атомов. (Например, органические соединения)
Хелаты – комплексы, содержащие полидентантные лиганды.
Комплексоны - полидентантные лиганды, содержащие несколько функциональных групп и образующие прочные комплексы практически со всеми двухзарядными ионами Me (Ca2+, Mg2+, Zn2+, Cu2+, Pt2+…)
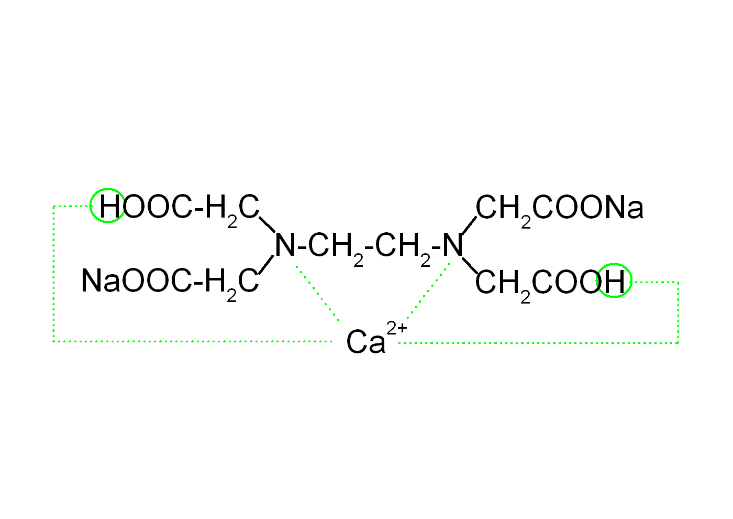 Трилон Б широко используется в клиническом анализе для титриметрического определения ионов Са2+ и Мg2+при определении жесткости воды. Максимальная дентатность такого лиганда равна 6.
Трилон Б широко используется в клиническом анализе для титриметрического определения ионов Са2+ и Мg2+при определении жесткости воды. Максимальная дентатность такого лиганда равна 6.
В краун-эфирах (особый тип полидентантных лигандов) донорные атомы кислорода заключены в плоский цикл определенного размера. Они содержат от 4 до 12 атомов кислорода (краун-4, краун-5 и т.д). Полости краун-эфиров имеют строго определенные размеры. Поэтому краун-эфиры могут избирательно связывать ионы металлов, размеры которых близки к размерам полости.
Значение:
Комплексные соединения – это ферменты, многие гормоны, лекарства, биологические-активные вещества. Комплексы белков с катионами металлов играют роль металлоферментов, катализирующих большинство химических превращений. Например, гормон инсулин, витамин B12, гемоглобин, хлорофилл. Также, КС составляют значительную часть природных материалов.
Комплексные соединения находят широкое применение:
- для выведения из организма камней, которые образуются в почках, мочевом пузыре, желчных протоках;
- для маскировки нежелательных примесных элементов в составе лекарственных препаратов;
- для очистки организма от ядов, радиоактивных элементов, тяжелых металлов;
- в аналитической химии для определения ионов, т.к. яркая окраска многих внутрикомплексных соединений позволяет использовать реакции их образования для обнаружения катионов металлов в анализируемом растворе;
- для разделения некоторых металлов и получения металлов высокой степени чистоты;
- для устранения жесткости воды;
- в качестве катализаторов важных биохимических процессов.
27. Понятие о ДЭС. Механизм образования. Термодинамика ДЭС. Уравнение Нернста, его анализ. Электродный потенциал. Стандартный электродный потенциал.
ДЭС можно разделить на 2 части: стабильную и подвижную. В стабильной части на поверхности металла возникает электрический заряд, который определяется потенциалопределяющими ионами (в рез-те электродных процессов). Подвижная часть содержит в себе 2 слоя. Адсорбционный, который состоит из противоионов, расположенных на расстоянии ионного радиуса от поверхности Me. (дейст. электростатические силы притяжения), и диффузный, который состоит из ионов, расположенных на расстоянии большем, чем ионный радиус. (свободно перемещаются в растворе, за счёт теплового движения).
ДЭС возникает в результате создания избытка носителей электричества данного знака по одну сторону и их недостатка по другую сторону границы раздела фаз, за счёт обмена заряженными частицами.
От величины электродного потенциала зависит величина работы, которая совершается системой при образовании ДЭС. Она выражается: А=n*F*Ф [кДж* 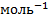 ]. В изобарно-изотермических условиях работа равна уменьшению энергии Гиббса A=-∆G.
]. В изобарно-изотермических условиях работа равна уменьшению энергии Гиббса A=-∆G.
Уравнение Нернста: 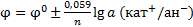 φ – электродный потенциал [В],
φ – электродный потенциал [В],
φ0 – стандартный электродный потенциал - характеризует природу электрода.
(φ = φ0, если a(п.о.и.) = 1 моль ·дм-3);
R – универсальная газовая постоянная,
n – число электронов в электродной реакции,
F – число Фарадея,
a(п.о.и.) – активная концентрация
потенциалопределяющих ионов [моль · дм-3].
Анализ: Электродный потенциал зависит от природы электрода, температуры и активной концентрации потенциалопределяющих ионов в жидкой фазе электрода.
Электродный потенциал – максимальная разность потенциалов, возникающая на границе раздела 2х фаз в момент установления равновесия.
Для измерения стандартного электродного потенциала 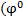 ) необходимо собрать гальваническую цепь из исследуемого электрода и стандартного водородного электрода, потенциал которого условно принят за 0В.
) необходимо собрать гальваническую цепь из исследуемого электрода и стандартного водородного электрода, потенциал которого условно принят за 0В.
28. Электроды сравнения. Стандартный водородный электрод, его устройство, электродная реакция, значение. Гальванические цепи для определения стандартных электродных потенциалов. Хлорсеребряный электрод, его электродная реакция, электродный потенциал.
Электроды сравнения – их потенциалы известны, постоянны и воспроизводимы. (Пр. хлорсеребряный)
Стандартный ВЭ состоит из черниной платиновой пластинки, насыщенной газообразным водородом при давлении 1 атм (101,3 кПа) и опущенной в раствор кислоты с активностью а( 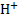 )= 1 моль*
)= 1 моль* 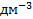 .
.
Электродная р-я: 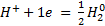 .
.
Значение возникающего электродного потенциала зависит от природы Me, концентрации ионов Me в растворе и от температуры.
Гальваническая цепь для определения стандартных электродных потенциалов состоит из исследуемого электрода и стандартного водородного электрода, потенциал которого условно принят за 0В.
ЭДС – разность потенциалов на концах гальванической цепи. ЭДС = Ф(+) – Ф(-).
Хлорсеребряный электрод представляет собой серебряную проволоку, покрытую слоем хлорида серебра и опущенную в насыщенный раствор хлорида калия.
Эл. Р.: 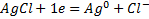
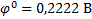
29. Электроды определения. Стеклянный электрод, его электродная реакция, электродный потенциал. Ионоселективные электроды и их применение в медико-биологических исследованиях. Потенциометрический анализ. Потенциометрическое титрование, сущность метода, значение для биологии и медицины.
Электроды определения – их потенциалы зависят от концентрации анализируемых ионов.
Стеклянный электрод состоит из стеклянной трубки, заканчивающейся шариком из специального стекла; внутрь этой системы наливают буферный раствор и для токоотвода помещают хлорсеребряный электрод. 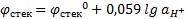 ;
; 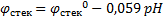 .
.
Ионоселективные электроды – это специальные электроды, равновесный потенциал которых в растворе электролита зависит от концентрации содержащихся в нём ионов. C их помощью можно наблюдать за изменением ионного состава биологических жидкостей в динамике, а также получать информацию о внутриклеточном изменении концентрации ионов Na+, К+, Сa2+, СI- и т.д.
Потенциометрический анализ – физико-химический метод, позволяющий определять концентрации ионов на основе измерения электродвижущей силы гальванической цепи, содержащей исследуемый раствор.
Потенциометрическое титрование – метод анализа, в котором точка эквивалентности определяется по изменению в ходе титрования электродвижущей силы гальванической цепи, содержащей анализируемый раствор. Методом потенциометрического титрования анализируют не только истинные растворы, но и мутные, окрашенные, коллоидные системы.
Метод потенциометрии позволяет объяснить механизм генерирования и распространения нервных импульсов, природу реакций биологического окисления и гидролиза. Метод прямой потенциометрии применяют для клинического анализа на ионы Н+, К+, Na+, Ca2+, Cl-, I-, HS- и других в биологических жидкостях, растворах лекарственных препаратов и солевых кровезаменяющих жидкостях, на молекулы глюкозы, мочевины, аминокислот, а также для контроля окружающей среды.
Межфазные явления их термодинамическая характеристика. Сорбция. Адсорбция. Абсорбция. Физическая и химическая сорбция. Понятие поверхностно активных веществ (ПАВ), их свойства и применение в медицине.
Поверхность раздела фаз – слой, возникающий на границе раздела фаз. (Т-Г, Т-Ж, T-T, Ж-Г, Ж-Ж)
Она характеризуется параметрами:
1. Удельная поверхность фазы S уд. S = 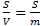 [
[ 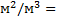 1/м или
1/м или 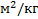 ]
]
2. Свободная поверхностная энергия Gs = σS [Дж(Н/ 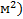 ]
]
3. Поверхностное натяжение σ = 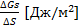
Поверхностная энергия подчиняется основным законам ТД:
Согласно первому закону ТД, поверхностная энергия может переходить в химическую, электрическую и свободную энергию Гиббса.
Согласно второму закону ТД, поверхностная энергия может быть причиной самопроизвольных процессов, определяемых уменьшением энергии Гиббса.
Сорбция – поглощение одного вещества другим. Сорбент – поглотитель. Сорбтив (сорбат) – поглощаемое вещество.
Адсорбция – поглощение поверхностью сорбента.
а = (Со – С) V / S, где
а – величина адсорбции (удельная сорбционная емкость) [ммоль м-2];
Со – начальная концентрация адсорбата [ммоль дм-3];
С – равновесная концентрация адсорбата [ммоль дм-3];
V – объем жидкой фазы [дм3];
S – площадь поверхности адсорбента [м2].
С увеличением концентрации адсорбируемого вещества величина адсорбции возрастает и достигает максимального значения при полном насыщении поверхности.
Математически эта взаимосвязь характеризуется уравнением Гиббса:
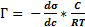 [ммоль м-2], где
[ммоль м-2], где
Г - количество адсорбированного вещества [ммоль/м2]
С – равновесная концентрация адсорбата [ммоль/л]
R – универсальная газовая постоянная
Т – абсолютная температура.
Величина адсорбции зависит от:
1. Размера поверхности адсорбента (↑ S ↑ Г).
2. Температуры (↑t ↓Г ).
3. Типа сорбента, его сродства к растворителю.
4. Заряда адсорбента и адсорбтива.
5. Концентрации адсорбтива.
Абсорбция – поглощение всем сорбентом.
Физическая сорбция происходит за счёт сил Ван-дер-Ваальса, и возникает только межмолекулярное взаимодействие
Химическая сорбция происходит за счёт химического взаимодействия и называется хемосорбцией.
Поверхностно-активные вещества (ПАВ) – вещества, обладающие низким поверхностным натяжением и вызывающие положительную адсорбцию. (Спирты, органические кислоты, сложные эфиры, белки, холестерол, жиры, липиды, мыла.)
Правило Траубе-Дюкло: При удлинении цепи на группу -СН2 - в гомологическом ряду способность к адсорбции возрастает в 3,2 раза.
Правило Ребиндера: На границе раздела полярные группы молекул ПАВ ориентируются в сторону более полярной фазы, а углеводородный радикал – в сторону менее полярной фазы.
ПАВ широко используются в фармации в качестве основы для приготовления мазей, свечей, эмульсий, а также солюбилизаторов.
Электролитная адсорбция, её закономерности. Правила электролитной адсорбции. Ионообменная адсорбция, её закономерности. Иониты, их классификация. Ионообменная ёмкость, способы её выражения. Применение ионитов в медицине.
Электролитная адсорбция – избирательная адсорбция ионов из раствора электролита на полярном адсорбенте.
Правила электролитной (избирательной) адсорбции:
1. Правило Панета-Фаянса: на поверхности кристалла преимущественно адсорбируются те ионы, которые входят в состав кристаллической решетки.
2. Правило изоморфизма: На полярном адсорбенте из раствора электролита преимущественно адсорбируются ионы, близкие по строению и размерам к одному из ионов кристаллической решетки адсорбента.
3. Если ионы-адсорбаты имеют одинаковые по знаку и разные по величине степени окисления, то в первую очередь адсорбируются ионы с большей степенью окисления: Fe 3+ > Ca 2+ > K + .
4. Если ионы-адсорбаты имеют одинаковые по величине и знаку степени окисления, то в первую очередь адсорбируются менее гидратированные ионы (с большим ионным радиусом).
Лиотропный ряд (ряд Гофмейстера) для катионов: Cs+ > Rb+ > K+ > Na+ > Li+; для анионов: SCN- > I- > Br- > Cl-.
Ионообменная адсорбция – процесс, при котором твёрдый адсорбент обменивает эквивалентное количество своих ионов на ионы того же знака из жидкого раствора.
Починяется закону эквивалентности, всем 4м правилам электронной адсорбции, принципу Ле-Шателье-Брауна, что позволяет регенерировать иониты.
Классификация ионитов:
По происхождению: природные (кристаллические силикаты, апатиты, гуминовые кислоты) и синтетические (алюмосиликаты, ионно-обменные смолы и высокомолекулярные вещества в качестве каркаса, целлюлоза)
По составу: неорганические (апатиты), органические (гуминовые кислоты, сапропель, целлюлоза)
В санитарно-гигиенической практике иониты используются для очистки воды, выделения и очистки радиоактивных изотопов, являются составной частью безотходных экологически чистых методов производства; для декальцинирования крови с целью ее консервации; для осуществления гемодиализа крови (используется ионит - алюмогель); беззондовой диагностики кислотности желудочного сока; детоксикации организма при различных отравлениях. Аниониты - антацидные средства, катиониты используются для предотвращения ацидоза, предупреждения и лечения отеков, связанных с декомпенсацией сердечной деятельности;
Мицелла и её строение. Электрокинетический или дзета-потенциал и его свойства. Зависимость агрегативной устойчивости мицеллы от величины дзета-потенциала и концентрации электролита. Изоэлектрические состояния мицеллы.
Мицелла – структурная единица коллоидных систем.
Пример.
AgNO3 + KI (избыток) = AgI + KNO3
Осадок AgI находится в избытке раствора KI.
Избыток электролита выполняет роль стабилизатора.
Осадок AgI яв-ся агрегатом мицеллы.
Ионы I- называются потенциалопределяющими.
Агрегат и потенциалопределяющие ионы составляют ядро мицеллы.
К отрицательному заряду будут притягиваться противоионы K+, образуя плотный слой противоионов.
Потенциалопределяющие ионы и противоионы плотного слоя вместе образуют адсорбционный слой.
Адсорбционный слой вместе с агрегатом составляют гранулу.
Часть противоионов, не вошедших в адсорбционный слой, образуют диффузный слой.
Гранула и диффузный слой составляют мицеллу.
Мицелла, таким образом, электронейтральна.
В мицелле выделяют две границы:
Граница раздела фаз - проходит между потенциалопределяющими ионами и противоионами плотного слоя.
Граница скольжения - проходит между гранулой и диффузным слоем.
На границе скольжения возникает электрокинетический потенциал или дзета (ξ) - потенциал.
Дзета-потенциал – один из основных факторов, определяющих поведение мицеллы в электрическом поле. Его рассчитывают по скорости движения частиц дисперсной фазы при электрофорезе или дисперсионной среды при электроосмосе.
Величина дзета-потенциала определяется толщиной диффузного слоя и зависит от разности между общим числом зарядов потенциалопределяющих ионов и числом зарядов противоионов, находящихся в адсорбционном слое.
Чем больше заряд гранулы и, соответственно, величина дзета-потенциала, тем устойчивее коллоидный раствор.
Наличие одноименного заряда способствует отталкиванию частиц друг от друга, препятствуя таким образом их коагуляции (укрупнению) и седиментации (осаждению).
Специфические свойства растворов ВМВ. Набухание. Степень набухания, факторы, влияющие на набухание. Ограниченное и неограниченное набухание. Вязкость. Удельная, приведённая и характеристическая вязкости. Осмотическое давление.
К специфическим свойствам растворов ВМВ относят: набухание, вязкость, осмотическое давление.
Набухание - это увеличение объема и массы полимера в результате избирательного поглощения низкомолекулярного вещества из жидкой или газообразной среды.
Степень набухания показывает отношение приращения объема или массы набухшего полимера к первоначальному объему или массе. Её определяют весовым и объемным методом. Весовым методом определяют массу сухого и набухшего полимера и по разности находят массу поглощенной жидкости. Объемным методом измеряют изменение объема тела при поглощении им жидкости, или объема жидкости, в котором происходит набухание. 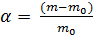 ;
; 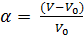
На степень набухания влияют:
1) Природа полимера и растворителя. Полярные ВМВ лучше набухают в полярных растворителях (например, белки в воде), неполярные в неполярных (например, каучук в бензоле).
2) Температура. Повышение температуры способствует более быстрому набуханию, так как усиливается движение частиц, что способствует разрыхлению внутренних структур. Для каждого высокомолекулярного вещества и растворителя должна существовать своя критическая температура, выше которой происходит их безграничное смешение.
3) Присутствие электролитов. Анионы способствуют набуханию в большей степени, чем катионы.
4) pH среды. Изменение рН среды в более кислую или щелочную сторону от изоэлектрической точки коллоида увеличивает степень набухания. Это объясняется появлением положительного и отрицательного заряда у коллоидных частиц и, следовательно, повышением степени гидратации.
Набухание называется ограниченным, если низкомолекулярная жидкость ограниченно растворима в ВМВ, и оно не заканчивается образованием текучей системы. Растянутая сетка макромолекул, стремясь сократиться, препятствует увеличению содержания растворителя.
При неограниченной растворимости низкомолекулярной жидкости в полимере его пачки после набухания продолжают раздвигаться и макромолекулы постепенно диффундируют в растворитель, образуя раствор. Такое набухание называют неограниченным.
Растворы высокомолекулярных соединений отличаются высокой вязкостью (или внутренним трением), обусловленной силами сцепления между молекулами жидкости. Количественно вязкость характеризуют коэффициентом вязкости η (этта), Па ∙ с.
Вязкое состояние обусловлено:
1) Силами сцепления гидрофильных молекул ВМВ с молекулами растворителя.
2) Образование ассоциатов при взаимодействии макромолекул между собой.
3) На аномально-высокую вязкость оказывает влияние форма и гибкость макромолекул полимера.
4) При протекании жидкости через сосуд отдельные части могут перемещаться с различными скоростями (гидродинамическая вязкость).
Удельная вязкость - увеличение вязкости, связанное с изменением концентрации при растворении полимера:
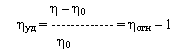 η – вязкость раствора;
η – вязкость раствора;
η0 – вязкость чистого растворителя;
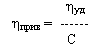 Для линейной (вытянутой) формы макромолекул удельную вязкость рассчитывают по уравнению Штаудингера: ηуд. = К· М(X) · C(X)
Для линейной (вытянутой) формы макромолекул удельную вязкость рассчитывают по уравнению Штаудингера: ηуд. = К· М(X) · C(X)
М (Х) – относительная молекулярная масса полимера [а. е. м.];
C (X) – весовая концентрация полимера [г · м-3];
К – константа, характеризующая особенности гомологического ряда полимера.
Величина ηуд/С получила название приведенной вязкости:
Предел ηуд/С при С→0 отражает гидродинамическое сопротивление движению молекул полимера и именуется характеристической вязкостью [η].
В растворах ВМВ осмотическое давление имеет ряд особенностей. Это связано с тем, что макромолекула ВМВ может рассматриваться как совокупность молекул меньшего размера.
Это учитывает уравнение Галлера: πосм = (СВМВ/МВМВ) ∙ RТ+βС2ВМВ, где
СВМВ – весовая концентрация полимера, г/м3;
β – коэффициент, учитывающий форму, гибкость, размеры макромолекулы.
Если концентрация раствора невелика, то βС2ВМВ → 0, тогда уравнение Галлера переходит в уравнение Вант-Гоффа. Измеряя осмотическое давление растворов различных концентраций и строя график зависимости πосм/ СВМВ от СВМВ, находят значение молярной массы полимера и коэффициента β.
Факторы, влияющие на осмотическое давление ВМВ:
Концентрация - с повышением концентрации ВМВ осмотическое давление возрастает.
Температура - при повышении температуры осмотическое давление возрастает.
pH - в изоэлектрической точке осмотическое давление будет минимальным, при смещении pH от изоэлектрической точки в кислую или щелочную области оно увеличивается.
Термодинамические функции состояния систем. Внутренняя энергия. Первый закон термодинамики, формулировка, математическое выражение, философское значение, применение к биологическим системам.
К термодинамическим функциям системы относятся:
1. Внутренняя энергия (U).
2. Энтальпия (Н).
3. Энтропия (S).
4. Энергия Гельмгольца (F).
5. Энергия Гиббса (свободная энергия) (G).
6. Химический потенциал (μ).
1. Внутренняя энергия (U) – складывается из кинетической энергии движения молекул или атомов, образующих систему, потенциальной энергии их взаимодействия и внутримолекулярной энергии. Абсолютное значение измерить невозможно, поэтому измеряют приращение:
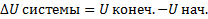
Внутренняя энергия есть функция состояния системы, приращение которой (ΔU) равно теплоте, поступающей в систему при изохорном процессе (V=const).
ΔU=Q V , где Q V - теплота изохорного процесса. ΔU - кДж/моль или кДж· моль-1
Первый закон термодинамики: Теплота, подведённая к системе, расходуется только на увеличение внутренней энергии системы и на совершение системой работы против внешних сил.
± Q = D U ± W, где Q – теплота получаемая или отданная системой, U – внутренняя энергия системы, W – работы, совершаемая системой или над системой.
В биологических системах теплота обычно отдается системой во внешнюю среду, а работа совершается за счет убыли внутренней энергии: - Q = ΔU - W.
Теплота (Q) – форма передачи энергии, посредством хаотического столкновения частиц соприкасающихся систем, системы и среды. Q = С· Δ T [Дж], где С - молярная теплоемкость [Дж моль/К].
Работа (W) – форма передачи энергии от системы в окружающую среду или другой системе, посредством упорядоченного взаимодействия частиц, вызванная преодолением сопротивления.
Первое начало термодинамики имеет огромное философское значение, поскольку, утверждая неуничтожимость энергии оно тем самым и утверждает неуничтожимость самой материи, т.к. энергия без материи существовать не может.
3. Термодинамические функции состояния системы. Энтальпия. Энтропия. Энергия Гиббса. Химический потенциал.
К термодинамическим функциям системы относятся:
1. Внутренняя энергия (U).
2. Энтальпия (Н).
3. Энтропия (S).
4. Энергия Гельмгольца (F).
5. Энергия Гиббса (свободная энергия) (G).
6. Химический потенциал (μ).
Энтальпия (Н) – позволяет оценить тепловой эффект реакции; это часть внутренней энергии системы, которая может совершить полезную работу. Это функция состояния системы, приращение которой равно теплоте, поступившей в систему в изобарном процессе. Абсолютное значение энтальпии измерить невозможно, поэтому измеряют ее приращение: DHсистемы = Hкон - Hнач ; ΔН = Ср ΔТ
Из первого закона ТД: Q = ΔU + W; Qр = ΔU + р·Δ V = (U2+р·V2) - (U1+ p·V1), где: Qр - теплота изобарного процесса при р=const; Н = U + р·V, где Н - энтальпия, Qр=Н2-Н1=ΔН, ΔН=Qр.
ΔН<0 – реакция идёт с выделением тепла, т.е. экзотермическая.
ΔН>0 – реакция идёт с поглощением тепла, т.е. эндотермическая.
Энтропия ( S) – мера неупорядоченности в системе. Характеризует связанную энергию. Это функция состояния системы, приращение которой (ΔS) равно минимальной теплоте Q, поступившей в систему в обратимом изотермическом процессе, делённой на абсолютную температуру (T), при которой совершается этот процесс. ΔS=Qmin/T, [Дж · моль-1 · К-1].
Энтропия связана с вероятностью состояния системы уравнением Больцмана:
S=КБ · InW, где KБ-постоянная Больцмана, KБ = R/Nа= 1,38· 10-23 Дж· К-1; W-вероятность состояния системы, т.е. число микросостояний, которым может быть реализовано данное макросостояние. Опытным путем определяют приращение энтропии: ΔS = S2 – S1.
ΔS<0 – снижение энтропии за счёт образования сложной структуры из более простых.
ΔS>0 – распад сложной структуры на более простые.
Свободная энергия Гельмгольца - термодинамический потенциал, убыль которого в изотермическом процессе равна работе, совершённой системой над внешними силами.
ΔF = ΔU – TΔS.
Энергия Гиббса ( G) - это часть потенциальной энергии реагирующих веществ, которая может быть использована для осуществления полезной работы.
В изобарно-изотермических условиях ΔG = ΔH – TΔS.
Анализ уравнения:
1 Энтальпийный фактор ΔH. Определяет стремление системы снизить свою энергию за счет образования сложных частиц из более простых, при этом совершается полезная работа.
2 Энтропийный фактор TΔ S. Определяет стремление системы к хаотичному неупорядоченному состоянию за счет распада сложных частиц на более простые и распределению их по всему объему системы.
Величина ΔG служит критерием возможности самопроизвольного протекания процессов.
Если ΔG<0 – процесс протекает самопроизвольно.
Если ΔG>0 – процесс самопроизвольно не протекает.
Если ΔG=0 – состояние равновесия.
Химический потенциал – часть свободной энергии, приходящаяся на 1 моль любого химического элемента при постоянных давлении, температуре и массе других веществ. Хим. потенциал какого-либо вещества в системе равен отношению энергии Гиббса (G) к количеству вещества (n). μ=G(x)/n(x), отсюда G(х)=n(х)·μ(х).
Для вещества, находящегося в растворе, μ зависит от концентрации раствора, и природы растворителя. Уравнение изотермы: μ(x) = μ0(x) + R·T ·In C(x), где μ(x)- химический потенциал [Дж · моль-1]; μ0(x)- стандартный химический потенциал; С(x) - молярная концентрация вещества x [моль · дм-3].
С увеличением концентрации вещества в системе μ увеличивается, т.е. ∆ μ(х)>0, а с уменьшением - снижается, т.е. ∆ μ(х)<0.
4. Термохимия. Термохимические уравнения, их особенности. Закон Гесса. Энтальпии образования и сгорания. Стандартные энтальпии образования и сгорания. Следствия из закона Гесса, формулировки, математические выражения, примеры.
Термохимия – раздел термодинамики, изучающий изменение энергии при протекании химических процессов.
Термохимическое уравнение – условное изображение физико-химического процесса.
Особенности:
1. Уравнения записываются с учетом термодинамических функций состояния системы (ΔH, Δ S).
2. Учитывается 1 моль вещества, поэтому возможны дробные коэффициенты.
3. Указываются агрегатные состояния веществ.
4. С термохимическими уравнениями могут производиться обычные алгебраические действия.
Пример: ½ N2(г)+ ½ O2(г)= NO(г),ΔH>0.
Закон Гесса: тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Следствия:
1. Тепловой эффект (энтальпия) реакции равен разности сумм теплот образования продуктов реакции и сумм теплот образования реагентов с учётом количества всех молей, участвующих в реакции.
Энтальпия образования: 
2. Тепловой эффект реакции равен разности сумм теплот сгорания реагентов и сумм теплот сгорания продуктов реакции.
Энтальпия сгорания: 
Стандартная энтальпия образования – образование 1 моля сложного вещества из простых при стандартных условиях.
Стандартная энтальпия сгорания – сгорание 1 моля вещества при стандартных условиях до образования высших оксидов.
Дата: 2019-04-23, просмотров: 691.