Уравнения Хартри-Фока для молекул можно, в принципе, решить численно, получив МО в виде таблиц. Извлечение, однако, химической информации из МО, представленных таким образом - нелегкая задача. Существует простое и мощное приближение, позволяющее существенно упростить как решение уравнений ХФ, так и интерпретацию результатов. Суть его в следующем.
Двигаясь по молекуле, каждый электрон попадает под преимущественное влияние поля ядра, вблизи которого электрон находится в данный момент. Это означает, что его МО вблизи этого ядра должна быть близкой к соответствующей АО. Поэтому каждую МО можно представить как линейную комбинацию всех АО системы:
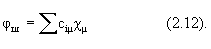
Коэффициенты разложения сim определяют “вес” каждой АО в МО; ясно, что одна и та же АО входит в разные МО с разными весами, т.е. эти коэффициенты различны для каждой МО.
Представление (2.12) очень удобно для применения вариационного метода. Применяя его с учетом ортонормированности МО, из условия минимума энергии получают уравнения ХФ в виде:
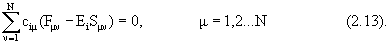
Элементы матрицы Фока в приближении МО ЛКАО имеют вид:
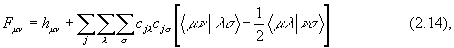
где
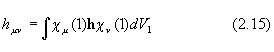
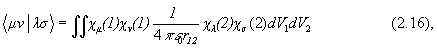
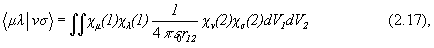
а Sij –матрица интегралов перекрывания между АО cm и cn , а Еm - одно из решений секулярного уравнения
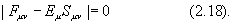
Уравнения (2.13) называются уравнениями Рутана.
Уравнения Рутана можно переписать в матричном виде:
F C = S C E , (2.19)
который путем унитарных преобразований F+=S-1/2FS-1/2 и C+=S-1/2C сводится к стандарнной задаче на собственные значения.
F+C+= EC+. (2.20)
Введем теперь матрицу P c элементами
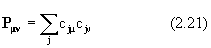
(суммирование ведется по занятым МО). Она называется матрицей зарядов-порядков связей или матрицей плотности; смысл этого названия будет объяснен позже. Эта матрица играет важную роль в теории химической связи, поскольку о писывает распределение электронной плотности по молекуле.
Энергия молекулы с закрытыми оболочками в терминах введенных обозначений записывается следующим образом:
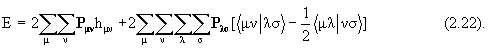
Решение уравнений Рутана осуществляется таким же итерационным методом ССП, как и в случае атома. Блок-схема вычислительного процесса представлена на рис. 2.1. 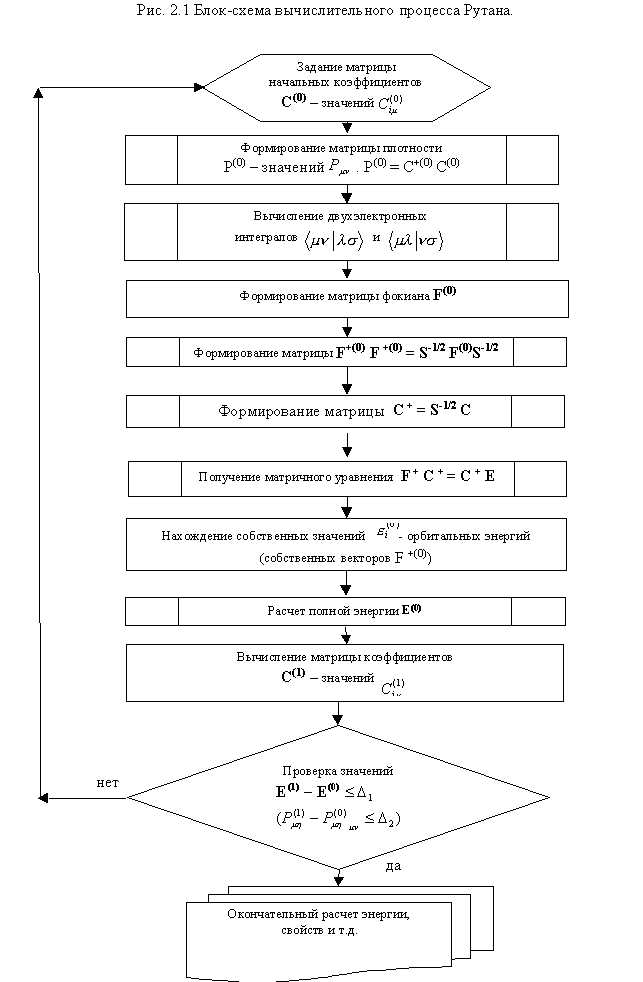
Двухэлектронные кулоновский 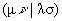 и обменный
и обменный 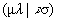 интегралы по АО (или по базисным функциям - см. ниже) являются причиной большинства практических проблем в случае, когда расчет проводится из первых принципов. Число их без учета симметрии равно » N4/8, где N - число АО, т.е. очень велико (для сравнения: число одноэлектронных интегралов hm n равно » N2/2 – см. табл. 2.2). Таким образом, вычисление этих интегралов и их запоминание в памяти компьютера представляет собой отдельную проблему, особенно, если учесть, что все четыре орбитали cm ,cn , cl и cs в общем случае центрированы на разных ядрах. Отсюда ясно, сколь важен для эффективного вычислительного процесса выбор аналитического вида функций, по которым вычисляются интегралы. В так называемых прямых ССП методах (доступных при наличии суперкомпьютеров), эти интегралы не запоминаются, а вычисляются в каждой итерации. При этом малыми по величине интегралами часто пренебрегают, что увеличивает риск ошибки, ибо число их велико, а знаки одинаковы (положительны).
интегралы по АО (или по базисным функциям - см. ниже) являются причиной большинства практических проблем в случае, когда расчет проводится из первых принципов. Число их без учета симметрии равно » N4/8, где N - число АО, т.е. очень велико (для сравнения: число одноэлектронных интегралов hm n равно » N2/2 – см. табл. 2.2). Таким образом, вычисление этих интегралов и их запоминание в памяти компьютера представляет собой отдельную проблему, особенно, если учесть, что все четыре орбитали cm ,cn , cl и cs в общем случае центрированы на разных ядрах. Отсюда ясно, сколь важен для эффективного вычислительного процесса выбор аналитического вида функций, по которым вычисляются интегралы. В так называемых прямых ССП методах (доступных при наличии суперкомпьютеров), эти интегралы не запоминаются, а вычисляются в каждой итерации. При этом малыми по величине интегралами часто пренебрегают, что увеличивает риск ошибки, ибо число их велико, а знаки одинаковы (положительны).
Таблица 2.2 Общее число одноэлектронных и двухэлектронных интегралов в зависимости от базиса
| Молекула | Базис | Одноэлектронные интегралы | Двухэлектронные интегралы | Общее число интегралов | |
| N | тип АО | ||||
| Н2 CH4 Бензол C6H6 | 2 9 36 | 1s 1s-, 2s-, 2p- АО углерода, 1s-AO водорода 1s-, 2s-, 2p-AO углерода, 1s-AO водорода | 3 45 666 | 6 1035 222111 | 9 1080 222777 |
Волновая функция и энергии, получаемые с помощью метода Рутана
(и с помощью метода Хартри-Фока вообще), инвариантны относительно ортогонального преобразования занятых электронами спин-орбиталей. Это означает, в частности, что если вместо АО для построения МО (2.12) будут использованы их линейные комбинации, полученные с помощью ортогональных преобразований, то одновременно и согласовано изменятся как все одно- и двухэлектронные интегралы, так и матрица P. Волновая функция (2.10) и энергия системы (2.22) при этом останутся прежними. Это очень важное свойство метода: используя его всегда можно облегчить концептуальную химическую трактовку результатов, например, перейти к локализованным МО, описывающим электроны связи и неподеленные электронные пары, гибридизованным АО, и т.д.
Ортогональным преобразованием называется преобразование, которое не меняет длин векторов и углов между ними и переводит один ортонормированный базис в другой ортонормированный. Матрица ортогонального преобразования Т обладает свойствами: ТТ' = 1, det T = ± 1.
Введение приближения МО ЛКАО ограничивает точность метода ХФ лишь постольку, поскольку конечное число АО реально включается в расчеты. В принципе, чтобы воспроизвести точное ХФ решение, потребовалось бы использовать бесконечное число функций в разложении (2.12) – этот случай известен как хартри-фоковский предел.
Приведем несколько примеров исследования свойств молекул неэмпирическим методом Хартри-Фока.
Таблица 2.3. Длины связи (А) и углы (град.) для циклофосфорамида
| связь | Рентгеновский Эксперимент | Расчет HF/6-31G* |
| P=O | 1.47 | 1.46 |
| P-O | 1.58 | 1.59 |
| P-N | 1.63 (экзоциклическая) 1.63 (эндоциклическая) | 1.65 (экзоциклическая) 1.67 (эндоциклическая) |
| C-Cl | 1.78, 1.79 | 1.79, 1.80 |
| - C-N-C | 117.0 | 117.7 |
| - P-N-C | 121.0 (экзоциклическая) 122.0 (эндоциклическая) | 119.7 (экзоциклическая) 122.4 (эндоциклическая) |
Таблица 2.4. Энергии (ккал/моль) вращательные барьеров
| молекула | STO-3G //STO-3G | 3-21G //3-21G | 6-31G* //6-31G** | эксперимент |
| BH3-NH3 | 2.1 | 1.9 | 1.9 | 3.1 |
| CH3-CH3 | 2.9 | 2.7 | 3.0 | 2.9 |
| CH3-NH2 | 2.8 | 2.0 | 2.4 | 2.0 |
| CH3-OH | 2.0 | 1.5 | 1.4 | 1.1 |
| CH3-SiH3 | 1.3 | 1.1 | 1.4 | 1.7 |
| CH3-PH2 | 1.9 | 1.7 | 2.0 | 2.0 |
| CH3-SH | 1.5 | 1.1 | 1.4 | 1.3 |
| Цис-HO-OH | 9.1 | 11.7 | 9.2 | 7.0 |
| Цис-HS-SH | 6.1 | 5.7 | 8.5 | 6.8 |
После символа // базис, с которым была оптимизирована геометрия.
Таблица 2.5. Энергии (ккал/моль) реакций изомеризации
| Формула | Реакция | HF/6-31G* // 3-21G | Эксперимент |
| HCN | Цианид водорода ® изоцианид водорода | 12.4 | 14.5 |
| CH2O | Формальдегид ® гидроксиметилен | 52.6 | 54.9 |
| CH3NO | Формамид ® нитрозометан | 65.3 | 62.4 |
| C2H3N | Ацетонитрил ® метил изоцианид | 20.8 | 20.9 |
| C2H4O |
Ограничения метода ХФ
В ряде молекулярных задач, решаемых методом ХФ, проявляется так называемая "дилемма симметрии". Дело в том, что из-за нелинейности уравнений ХФ среди решений всегда имеются такие, симметрия которых отличается от симметрии ядерной конфигурации молекулы. Класс однодетерминантных функций, обладающих надлежащей симметрией, всегда уже, чем при отсутствии симметрийных ограничений. Последние же дают более низкое значение энергии из-за дополнительной вариационной свободы. Таким образом, в вариационной процедуре возникает дилемма: что лучше - более низкая энергия или правильная симметрия орбиталей? Пример: Для правильного шестиугольника Н6 при больших расстояниях между атомами существуют решения, обладающие симметрией относительно 3-го, а не 6-го порядка. Такая же ситуация встречается в полиенах.
Метод ХФ также непригоден для расчета энергии Ферми в металлах.
Ограничения метода ХФ
В ряде молекулярных задач, решаемых методом ХФ, проявляется так называемая "дилемма симметрии". Дело в том, что из-за нелинейности уравнений ХФ среди решений всегда имеются такие, симметрия которых отличается от симметрии ядерной конфигурации молекулы. Класс однодетерминантных функций, обладающих надлежащей симметрией, всегда уже, чем при отсутствии симметрийных ограничений. Последние же дают более низкое значение энергии из-за дополнительной вариационной свободы. Таким образом, в вариационной процедуре возникает дилемма: что лучше - более низкая энергия или правильная симметрия орбиталей? Пример: Для правильного шестиугольника Н6 при больших расстояниях между атомами существуют решения, обладающие симметрией относительно 3-го, а не 6-го порядка. Такая же ситуация встречается в полиенах.
Метод ХФ также непригоден для расчета энергии Ферми в металлах.
Дата: 2019-02-19, просмотров: 410.