До этого момента мы игнорировали тот факт, что электроны относятся к классу фермионов - частиц с полуцелым спином (собственным моментом количества движения). Спин электрона равен ± 1/2 (в единицах 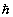 ). В квантовохимической литературе два возможных значения спина электрона a и b обозначаются символами
). В квантовохимической литературе два возможных значения спина электрона a и b обозначаются символами 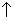 и
и 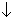 . Часто говорят "спин вверх" или "спин вниз", чтобы указать разные значения спина электрона. Чтобы учесть наличие спина, в аргумент одноэлектронных функций вводят спиновую переменную s, а функцию c i (xi) называют спин-орбиталью (xi=ri,si).
. Часто говорят "спин вверх" или "спин вниз", чтобы указать разные значения спина электрона. Чтобы учесть наличие спина, в аргумент одноэлектронных функций вводят спиновую переменную s, а функцию c i (xi) называют спин-орбиталью (xi=ri,si).
Из-за наличия спина имеются специальные ограничения на электронную волновую функцию, известные как принцип Паули.
1. Электронная волновая функция системы из нескольких электронов должна быть антисимметричной (менять знак) относительно обмена положениями и спинами пары из любых двух электронов i и j:
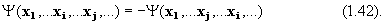
2. Никакие два электрона не могут быть в одном и том же квантовом состоянии (принцип исключения).
Чтобы понять, как нужно строить волновую функцию с учетом последнего обстоятельства, рассмотрим двухэлектронный атом гелия. Две эквивалентных волновых функции Хартри для этой системы имеют вид (1.24):

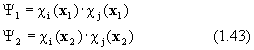
Очевидно, что ни одна из них не является антисимметричной. Однако легко заметить, что связанная с ними функция
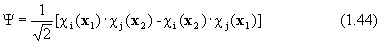
антисимметрична, 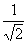 – нормировочный коэффициент.
– нормировочный коэффициент.
С математической точки зрения, вид этой волновый функции есть детерминант из спин-орбиталей c i(х):
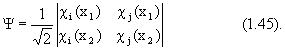
Если же мы попытаемся поместить электроны 1 и 2 на одну и ту же спин-орбиталь c i, то получим:
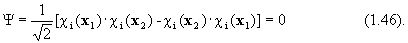
Значит волновая функция в виде (1. 44) удовлетворяет принципу исключения Паули.
Важным свойством детерминанта, известным из курса высшей математики, является то, что он меняет знак при перестановке двух любых его столбцов или строк; величина его при этом остается неизменной. Это как раз эквивалентно перемене мест двух электронов: электрон 1 перемещается с орбитали i на орбиталь j, а электрон 2 - с орбитали j на орбиталь i. Если же два столбца или две строки детерминанта одинаковы (что эквивалентно занятию двумя электронами одной и той же спин-орбитали), детерминант равен нулю.
Принцип Паули диктует, что две спин-орбитали с одинаковыми пространственными частями (т.е. с одинаковыми квантовыми числами n, l, m) отличаются только спиновыми компонентами. Пренебрегая малым по величине спин-орбитальным взаимодействием, каждую спин-орбиталь ci(x) можно представить в виде произведения пространственной орбитали ci(r) и спиновой функции h(s):ci(x)=ci(r)h(s).
На примере атома Не рассмотрим основное (синглетное) состояние. Из возможных вариантов распределения электронов между 1s и 2s – АО атома Не (рис. 7) этот описывается схемой а). 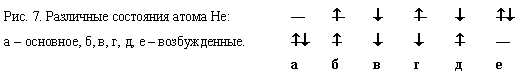
Электрон со спиновой функцией a обзначен стрелкой, направленной вверх, b - вниз.
Волновая функция основного состояния, выраженная через определитель, запишется так:
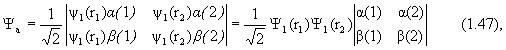
где функция 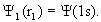
Y a имеет симметричную пространственную часть и антисимметричную спиновую. Полный спин системы равен нулю, поэтому это состояние принято называть синглетным.
Заполнение орбиталей электронами происходит в порядке возрастания их энергий согласно принципу Паули и каждая не пустая АО может быть занята либо одним, либо двумя (но не более) электронами, причем в последнем случае электроны обязательно имеют противоположные спины и называются спаренными. АО с одинаковыми квантовыми числами n и l заполняются так, чтобы суммарный спин электронов был максимален (правило Хунда); другими словами сначала заполняется максимальное число АО с разными m.
Для нейтральных атомов в основном состоянии последовательность заполнения АО следующая:
1s < 2s < 2p < 3s < 3p < 4s ~ 3d < 4p < 5s ~ 4d < 5p < 6s ~5d ~ 4f < 6p < 7s < ...
Это ведет к так называемому aufbau-принципу, который служит обоснованием таблицы Менделеева. Следует подчеркнуть, что порядок заполнения орбиталей обусловлен не отношением их энергий, а требованием минимума полной энергии атома, которая отлична от суммы одноэлектронных энергий (см. выше). Здесь также играют роль спин-орбитальные и другие эффекты. Именно поэтому, в частности, 4s-AO заполняются электронами в атомах К и Ca раньше, чем 3d АО.
В итоге электроны с одним и тем же значением главного квантового числа n образуют электронные слои (оболочки). Эти слои, в свою очередь, построены из подоболочек, заполненных электронами с одинаковым значением орбитального квантового числа l. Слои с n=1,2,3, 4 ... обозначают буквами K, L, M, N,...
Детерминант Слейтера
Итак, представление многоэлектронной волновой функции в виде детерминанта обеспечивает ее правильные антисимметричные свойства. Кроме того, электроны неразличимы и их перестановка не должна менять свойства системы. Перестановка электронов для детерминанта эквивалентна перестановке местами столбцов (строк), что лишь меняет знак детерминанта. Поскольку волновая функция в принципе определена с точностью до фазового множителя, перемена знака свойств системы не меняет. Приближенная многоэлектронная волновая функция, построенная из ортонормированных спин-орбиталей отдельных электронов, называется детерминантом Слейтера:
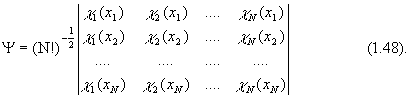
Детерминант Слейтера является единственной функцией, обеспечивающей антисимметричность волновой функции в орбитальном приближении. Следовательно, он дает только одно решение соответствующих одноэлектронных уравнений.
Хотя электроны неразличимы, в орбитальном приближении каждый электрон описывается "своей" волновой функцией. Системы, в которых все электроны спарены на орбиталях, называются системами с закрытыми (или замкнутыми) электронными оболочками. Для таких систем детерминант Слейтера состоит из дважды занятых электронами спин-орбиталей, число которых равно половине числа электронов. Системы с нечетным числом электронов называются системами с открытыми (незамкнутыми) оболочками.
Метод Хартри-Фока
Аппроксимация многоэлектронной волновой функции единственным детерминантом Слейтера (1.48) и использование приближения самосогласованного поля приводят к методу Хартри-Фока (ХФ). При этом исходное электронное уравнение Шредингера (1.20) путем довольно громоздких математических вычислений (см., например, М.Дьюар. Теория молекулярных орбиталей в органической химии) преобразуется в уравнения, где точный гамильтониан H (1.20) заменен оператором Фока (фокианом):
F = 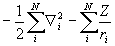 +
+ 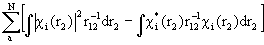 (1.49)
(1.49)
(Здесь и далее мы используем принятую в квантовой химии атомную систему единиц: множитель 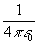 опускается, m = 1, e = 1, =1. Введение атомных единиц делает формулы менее громоздкими).
опускается, m = 1, e = 1, =1. Введение атомных единиц делает формулы менее громоздкими).
Различие между F и H состоит в том, что оператор кулоновского межэлектронного взаимодействия 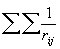 заменен оператором в квадратных скобках, описывающим взаимодействие каждого электрона со средним полем всех остальных электронов с учетом требований принципа Паули. Из условия минимума энергии
заменен оператором в квадратных скобках, описывающим взаимодействие каждого электрона со средним полем всех остальных электронов с учетом требований принципа Паули. Из условия минимума энергии 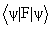 возникает набор независимых уравнений для каждой одноэлектронной орбитали - уравнения Хартри-Фока:
возникает набор независимых уравнений для каждой одноэлектронной орбитали - уравнения Хартри-Фока:
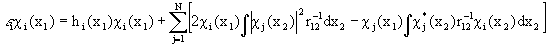 1.50
1.50
Энергия электрона, находящегося на орбитали 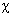 i, может быть получена умножением слева выражения (1.50) на i и интегрированием по всему пространству:
i, может быть получена умножением слева выражения (1.50) на i и интегрированием по всему пространству:
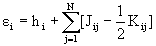 (1.51),
(1.51),
где 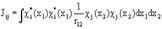 , (1.52),
, (1.52),
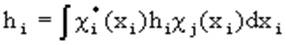 (1.53),
(1.53),
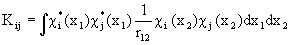 . (1.54)
. (1.54)
Одноэлектронный интеграл 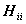 описывает энергию электрона на орбитали
описывает энергию электрона на орбитали 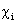 в поле ядра без остальных электронов.
в поле ядра без остальных электронов.
Двухэлектронный кулоновский интеграл 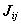 описывает энергии межэлектронного отталкивания при независимом движении электронов.
описывает энергии межэлектронного отталкивания при независимом движении электронов.
Двухэлектронный обменный интеграл 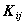 отражает понижение энергии электронов с параллельными спинами на орбиталях и
отражает понижение энергии электронов с параллельными спинами на орбиталях и 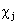 .
.
Полная энергия атома с замкнутыми оболочками (по 2 электрона на каждой орбитали) вычисляется в методе ХФ следующим образом:
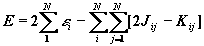 (1.55)
(1.55)
Подчеркнем, что оператор Фока сам зависит от полного набора одноэлектронных волновых функций и его решение ищется самосогласованно. По этой причине метод ХФ иногда отождествляют с методом ССП. Поскольку, однако, общая стратегия самосогласования проявляется в квантовой химии во многих контекстах, название "метод Хартри-Фока" более точно.
Оператор Фока состоит из трех членов:
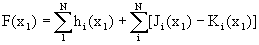 (1.56)
(1.56)
Здесь hi - точный одноэлектронный оператор (1.22):
hi = - 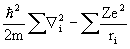 (1.57)
(1.57)
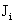 - кулоновский оператор:
- кулоновский оператор:
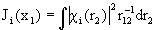 (1.58)
(1.58)
и 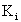 - нелокальный обменный оператор:
- нелокальный обменный оператор:
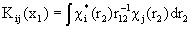 (1.59)
(1.59)
Наличие обменного члена в методе ХФ эквивалентно введению поправки на корреляцию движения электронов, описываемых разными орбиталями. Другими словами, этим мы учитываем корреляцию между движением электронов с разными спинами (обменную корреляцию). Кулоновская корреляция, вызванная взаимным отталкиванием электронов, независимо от их спинов, в методе ХФ не учитывается: она является следствием приближения независимых частиц. Это - существенный недостаток метода и мы к нему еще вернемся. Кроме того, в противоположность точной волновой функции, однодетерминантная функция ХФ вследствие самосогласования не имеет сингулярности при |ri - rj|® 0, следующей из (1.6).
Уравнения ХФ могут, в принципе, быть решены численно любым стандартным методом решения интегро-дифференциальных уравнений (например, методом Монте Карло, методом сетки и др.).
Дата: 2019-02-19, просмотров: 533.