 |
Почему же в промежуточных реакциях могут быть более высокие константы скорости? Прежде чем ответить на этот вопрос, для произвольной двусубстратной реакции вида
| |
запишем уравнение скорости, используя формулу Эйринга (18.40):
Отсюда можно сформулировать три принципиальных способа ускорения реакций
1. Первый из них — увеличение эффективной концентрации реагентов.
а) Данный механизм явно отсутствует в приведенных двух примерах катализа низкомолекулярными веществами (ионами Fe2+ и молекулами I2).
Но он может играть важную роль, когда поверхность катализатора достаточно велика и на ней предварительно связываются молекулы реагентов. Такая ситуация имеет место при гетерогенном катализе, а также при ферментативном катализе, осуществляемом макромолекулами (каковыми являются белки).
в) Так вот, при связывании молекул реагентов с катализатором могут происходить их сильное сближение друг с другом и правильная ориентация друг относительно друга, что и означает повышение эффективной концентрации.
2. Второй принципиальный способ ускорения касается энергии активации.
 |
а) Вспомним её определение (п. 18.2):
т.е. это разница между средней и барьерной (в отношении реакционной активности) энергией вещества.
б) Отсюда второй способ ускорения — это уменьшение энергии активации
«снизу», т.е. за счет увеличения средней энергии реагентов ( 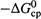 ; рис. 20.1).
; рис. 20.1).
Этот способ тоже возможен лишь в случае катализаторов с достаточно
большой поверхностью (ферменты; гетерогенный катализ).
в) Можно представить, что связывание реагента с катализатором сопровождается созданием напряжения в молекуле реагента. Из-за взаимодействия с химическими группами катализатора, определенная связь в реагенте как бы растягивается» (ослабевает), что означает переход реагента в возбуждённое состояние.
г) Когда же превращение молекулы реагента в продукт завершается, часть высвобождающейся в реакции энергии идёт на восстановление прежней структуры катализатора. Таким образом, повышение энергии реагента катализатором совершается как бы «в долг» – в счёт части энергии последующей реакции.
 |
3. а) И, наконец, третий способ ускорения — уменьшение энергии активации «сверху», т.е. путем снижения энергетического барьера ( 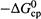 ; рис. 20.2).
; рис. 20.2).
б) Этого можно достичь путем разбиения исходной реакции на несколько других с меньшими значениями барьерной энергии.
 |
Действительно, допустим, что реакция (20.23) заменяется на две другие:
Если соответствующие группы катализатора уже находятся в реакционно-
активном состоянии, то на первой стадии необходимо активирование лишь
реагента S1, а на второй — реагента S2.
в) В исходной же реакции требуется одновременная активация сразу обоих реагентов. Следовательно, энергия активации каждой промежуточной реакции ниже (за счет более низкого барьера) энергии активации исходной реакции. И наиболее высокое из этих небольших значений 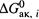 определяет константу скорости результирующего превращения.
определяет константу скорости результирующего превращения.
г) Очевидно, именно данный способ реализуется при катализе неорганическими веществами. В случае же ферментов могут использоваться все три способа (хотя, конечно, не обязательно, что все способы — сразу).
4. Продемонстрируем, насколько эффективно влияет снижение ∆G0ак на константу скорости. Пусть под влиянием катализатора ∆G0ак уменьшается от 109 кДж/моль до 74,5 кДж/моль.
 |
Тогда, согласно формуле Эйринга (18.44), при t = 37°С
т.е. скорость реакции увеличивается в миллион раз.
Уравнение Михаэлиса-Ментен
Теперь обратимся к кинетике каталитических реакций. Для определенности, будем иметь в виду ферментативные реакции, хотя многое из нижеследующего справедливо и для прочих каталитических процессов.
 |
1. Как и для цепных реакций, уравнение скорости ферментативного процесса зависит от его конкретного механизма. Рассмотрим простейший случай: односубстратныв необратимые реакции вида
протекающие в две стадии:
 |
На первой (обратимой) стадии образуется фермент-субстратный комплекс ES , а на второй он необратимо распадается на фермент и продукт реакции.
2. Вывод уравнения скорости производится по тому же принципу, что и для
цепных реакций (п. 20.2).
а) Так, тоже исходят из условия стационарности. В данном случае оно означает постоянство концентрации комплекса ES :
 |
б) Заметим, что концентрация свободного фермента равна
 |
где c E,0 — общая концентрация фермента — и в свободном состоянии, и в составе комплекса ES .
 |
в) Подставляя c E в (20.29), можно найти стационарную концентрацию комплекса:
 |
Здесь введена константа Михаэлиса:
 |
г) Скорость же образования конечного продукта, а значит и скорость реакции в целом, очевидно, такова:
 |
д) Подставляя c ES, получаем искомое выражение:
Величина Vmax — это максимальная скорость реакции; формула же (20.34, а) — уравнение Михаэлиса—Ментен, являющееся основным в ферментативной кинетике.
3. Проанализируем это уравнение.
 |
а) При малых концентрациях субстрата S (когда c S « KM) имеем:

т.е. фермент работает в линейном режиме, а реакция имеет первый порядок.
б) Если же, напротив, концентрация субстрата очень велика, получаем режим насыщения:

Здесь скорость перестает зависеть от концентрации субстрата, не поднимаясь выше Vmax, т.е. реакция приобретает нулевой порядок.
в) Отсюда — та гипербола, которой описывается зависимость v от 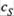 для ферментативной реакции (рис. 20.3). По мере же расходования субстрата порядок реакции изменяется от нулевого до первого.
для ферментативной реакции (рис. 20.3). По мере же расходования субстрата порядок реакции изменяется от нулевого до первого.
Дата: 2019-02-02, просмотров: 403.