Институт материаловедения и технологий материалов
Кафедра «Технология композиционных материалов, конструкций
И микросистем»
КУРСОВАЯ РАБОТА
По дисциплине: Теория гетерогенных сред
_____________________________________________________________________________
На тему: Диэлектрические свойства тонких полимерных пленок и их изменения в процессе сорбции воды
Студент гр. 1НАН-4ДБ-043 _______________________/ Ткаченко Т.П. /
(подпись) (Фамилия, имя, отчество)
Руководитель _____________________________________/ Козлов Н.А. /
(подпись) (Фамилия, имя, отчество)
К з а щ и т е д о п у с т и т ь
Зав. кафедрой ТКМКМ /Бабаевский П.Г./
(подпись) (Фамилия, имя, отчество)
“___”__________________ 20____г.
Оценка ______________________ Дата защиты __________________
Председатель комиссии _______________________/___________________/
(подпись) (Фамилия, имя, отчество)
МОСКВА – 2018
Содержание
Введение. 3
1 Диэлектрические свойства полимеров. 4
1.1 Основные представления о диэлектрических свойствах. 4
1.2 Феноменологическая теория диэлектрических свойств полимеров. 9
1.2.1 Основные уравнения. 9
1.2.2 Диэлектрические свойства системы, имеющей одно время релаксации. 12
1.2.3 Зависимость диэлектрических свойств полимеров от температуры.. 13
1.3 Влияние низкомолекулярных веществ на диэлектрические свойства полимеров. 14
2 Теоретические основы сорбции низкомолекулярных полимерами. 18
2.1 Адсорбция газов и паров. 18
2.2 Равновесная абсорбция. 21
2.3 Диффузия воды полимерами. 27
3 Влияние влаги на диэлектрические свойства различных полимерных пленок. 29
3.1 Изменение диэлектрических свойств тонкой полиамидной пленки. 29
3.2 Изменение диэлектрических свойств тонких пленок гибко- и жесткоцепных полимеров 32
3.3 Изменение диэлектрической проницаемости адсорбированной воды в полиимидных пленках 36
Заключение. 39
Список литературы.. 40
Введение
Тенденцией развития современных приборов, потребительских товаров и их узлов является их миниатюризация. Часто в основе конструкции различных плат, датчиков, актюаторов и прочих объектов техники используются тонкие полимерные пленки, выполняющие роль различных функциональных слоев и которые определяют работоспособность электронных устройств. При важно знать диэлектрические свойства таких пленок и зависимость этих свойств от влажности окружающей среды. В связи с вышеизложенным работа посвящена рассмотрению диэлектрические свойств тонких полимерных пленок и их изменению в процессе сорбции воды. Актуальность темы обусловлена широким применением тонких полимерных пленок в микросистемной технике в качестве диэлектрических, а так же сорбирующих слоев.
Цель курсовой работы состоит в рассмотрении диэлектрических свойств полимеров, теоретических основ явления сорбции и особенностей изменения диэлектрических свойств тонких полимерных пленок в процессе сорбции низкомолекулярных веществ.
Для достижения поставленной цели в работе необходимо решить следующие задачи:
1. Рассмотреть основные диэлектрические свойства полимерных пленок – поляризацию, электрическое смещение, время диэлектрической релаксации, диэлектрическую проницаемость;
2. Проанализировать феноменологическую теорию – один из подходов описания диэлектрических свойств, который включает в себя описание макроскопических свойств и характеристик диэлектрика;
3. Описать теоретические основы сорбции и диффузии низкомолекулярных веществ полимерами;
4. Собрать и обобщить данные о влияние сорбции низкомолекулярных веществ, в т.ч. влаги на диэлектрические свойства тонких полимерных пленок.
В соответствии с поставленными в работе задачами она состоит из трех глав.
В первой главе рассматриваются диэлектрические характеристики, такие как поляризация единицы объема диэлектрика, дипольный момент, диэлектрическая восприимчивость, зависимость диэлектрической проницаемости и диэлектрических потерь от частоты электрического поля, температуры. Так же описаны особенности изменения диэлектрических свойств полимеров в процессе сорбции.
Во второй главе рассмотрена теория сорбции, в частности адсорбция в макропористых полимерах и абсорбция паров непористыми полимерами, отдельно рассмотрен процесс диффузии низкомолекулярных веществ полимерами.
В третьей главе проанализированы изменения диэлектрических свойств под действием сорбированной пленками воды на примере четырех различных тонких полимерных пленок на основе полиамида, полиимида, поливинилового спирта и отвержденной алифатическим амином смолы ЭД-20. Так же рассматриваются диэлектрические характеристики (дипольный момент, диэлектрическая проницаемость) самой сорбированной воды.
Основные уравнения
Рассмотрим связь между электрическим смещением D и напряженностью электрического поля в полимерном диэлектрике.
Электрическое смещение в изотропной среде может быть представлено в виде:

где 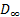 - электрическое смещение в диэлектрике, соответствующее случаю, когда частота электрических колебаний
- электрическое смещение в диэлектрике, соответствующее случаю, когда частота электрических колебаний 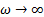 ;
; 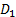 – «диссипативная» часть электрического смещения.
– «диссипативная» часть электрического смещения.
Электрическое смещение 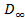 имеет вид, представленный в уравнении (1.15). Диссипативная же часть отличается от только значением диэлектрической проницаемости диэлектрика, которое имеет значение для
имеет вид, представленный в уравнении (1.15). Диссипативная же часть отличается от только значением диэлектрической проницаемости диэлектрика, которое имеет значение для 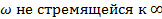 , а имеющей конкретное значение.
, а имеющей конкретное значение.
Представление электрического смещения в виде суммы двух членов является достаточно обоснованным, так как электрический момент диэлектрика, возникающий при наложении на него электрического поля, имеет две составляющие, одна из которых (деформационная) устанавливается очень быстро ( 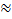 10-13 с.) [1]
10-13 с.) [1]
Следует заметить, что выражения для электрического смещения D типа (  15) справедливы лишь для стационарных электрических полей. Если электрическое поле Е, приложенное к диэлектрику, периодически изменяется во времени, то электрическое смещение в каждой точке среды в данный момент времени будет зависеть не только от напряженности электрического поля и скорости его изменения, но и от предыстории среды. Если время изменения электрического поля становится соизмеримым с временем установления статистического равновесия, то в среде активную роль будут играть релаксационные процессы. [1] Переход к равновесию может происходить таким образом, что будет иметь место суперпозиция релаксационных процессов. В связи с этим в реальном полимерном диэлектрике, находящимся в периодически изменяющемся электрическом поле, вектор электрического смещения должен каким-то образом учитывать предысторию среды.
15) справедливы лишь для стационарных электрических полей. Если электрическое поле Е, приложенное к диэлектрику, периодически изменяется во времени, то электрическое смещение в каждой точке среды в данный момент времени будет зависеть не только от напряженности электрического поля и скорости его изменения, но и от предыстории среды. Если время изменения электрического поля становится соизмеримым с временем установления статистического равновесия, то в среде активную роль будут играть релаксационные процессы. [1] Переход к равновесию может происходить таким образом, что будет иметь место суперпозиция релаксационных процессов. В связи с этим в реальном полимерном диэлектрике, находящимся в периодически изменяющемся электрическом поле, вектор электрического смещения должен каким-то образом учитывать предысторию среды.
Поэтому при описании диэлектрических свойств полимерных диэлектриков должны использоваться некоторые неравновесные значения 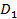 , отличающиеся о стационарных значений D01, соответствующих постоянным во времени электрическим полям. [1]
, отличающиеся о стационарных значений D01, соответствующих постоянным во времени электрическим полям. [1]
Полагая, что отклонение от стационарного значения 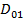 не велико, можно написать следующее релаксационное уравнение:
не велико, можно написать следующее релаксационное уравнение:

Уравнение ( 22) можно рассматривать как следствие фундаментальных положений термодинамики необратимых процессов. Если электрическое смещение стационарно, то 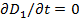 и
и 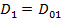 . Параметр
. Параметр 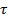 представляет собой время диэлектрической релаксации.
представляет собой время диэлектрической релаксации.
В случае периодических гармонических процессов уравнение ( 23) принимает вид

Отсюда следует, что:

Из сопоставления (24) и (15) становится очевидным, что диэлектрическая проницаемость 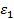 может быть представлена в комплексной форме:
может быть представлена в комплексной форме:

где 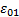 – «статическое» низкочастотное значение диэлектрической проницаемости.
– «статическое» низкочастотное значение диэлектрической проницаемости.
Реальные полимерные диэлектрики обычно описываются не одним временем релаксации, а спектром времен релаксации. Существует несколько причин, приводящих к появлению спектра времен релаксации. К их числу относятся: неодинаковая скорость протекания релаксационных процессов на различных участках тела и наличие различных релаксационных механизмов. [1] В случае полимеров к появлению спектров приводит уже само наличие длинных полимерных цепей и специфика межмолекулярного взаимодействия.
В случае произвольного числа релаксационных процессов выражение ( 25) может быть представлено в виде:

Таким образом, учитывая ( 26), и ( 20),(1,15), ( 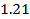 ), получаем:
), получаем:
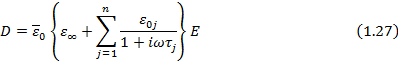
Выражение, стоящее в скобках, можно рассматривать как оператор диэлектрической проницаемости:

Этот оператор может быть представлен в дифференциальной форме:

Сопоставляя выражения ( 17), ( 18), ( 20) и ( 27) и разделяя действительную и мнимую части в ( 26), получаем следующие выражения для 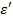 и
и 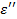 :
:


Эти формулы указывают на наличие дискретного спектра времен диэлектрической релаксации, возникающего в полимере, находящемся в переменном электрическом поле. Переходя от дискретного спектра к сплошному, при 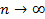 из уравнений ( 30) и ( 31) получим:
из уравнений ( 30) и ( 31) получим:


где 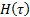 представляет собой плотность спектра времен диэлектрической релаксации.
представляет собой плотность спектра времен диэлектрической релаксации.
Рассмотрим случай 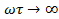 . Тогда
. Тогда 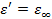 , а
, а 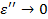 . Для полимеров это условие соответствует или очень высоким частотам (
. Для полимеров это условие соответствует или очень высоким частотам ( 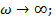 . =const ), или очень низким температурам(
. =const ), или очень низким температурам( 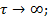 .
. 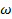 =const). Значит, из феноменологической релаксационной теории следует, что при низких температурах можно ожидать уменьшения диэлектрической проницаемости и соответствующих потерь .
=const). Значит, из феноменологической релаксационной теории следует, что при низких температурах можно ожидать уменьшения диэлектрической проницаемости и соответствующих потерь .
В другом предельном случае, когда 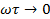 , что соответствует низким частотам или сравнительно высоким температурам, , а возрастает, достигая значения:
, что соответствует низким частотам или сравнительно высоким температурам, , а возрастает, достигая значения:

Адсорбция газов и паров
Адсорбция – это явление самопроизвольного концентрирования молекул одного вещества (адсорбата) на границе раздела фаз с другим веществом (адсорбентом). Происходит это вследствие уменьшения свободной поверхностной энергии в поверхностном слое (на границе раздела фаз) по сравнению с ее значением в объеме фазы. Адсорбционное уравнение Гиббса описывает это явление:

где 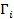 – избыток i-го компонента (адсорбата) в поверхностном слое; dμi - изменение химического потенциала (парциальной мольной свободной энергии) i-го компонента при адсорбции;
– избыток i-го компонента (адсорбата) в поверхностном слое; dμi - изменение химического потенциала (парциальной мольной свободной энергии) i-го компонента при адсорбции; 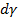 – изменение свободной поверхностной энергии.
– изменение свободной поверхностной энергии.
Данное уравнение выводится из обобщенного уравнения Гиббса-Дюгема для многокомпонентных гетерогенных систем (объемных фаз). Нас же интересует случай, когда количество компонентов системы ограничивается двумя. [5]Тогда уравнении Гиббса записывается в виде:

где 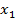 и
и 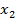 - мольные доли компонентов.
- мольные доли компонентов.
Величины 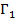 и
и 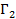 зависят от выбора разделяющей поверхности на границе раздела фаз. Общее изменение удельной свободной поверхностной энергии при адсорбции называется поверхностным давлением
зависят от выбора разделяющей поверхности на границе раздела фаз. Общее изменение удельной свободной поверхностной энергии при адсорбции называется поверхностным давлением 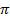 и представляет из себя разность энергий до и после адсорбции (2.3).
и представляет из себя разность энергий до и после адсорбции (2.3).

При контакте поверхности полимеров с газами или парами низкомолекулярных летучих веществ при заданных Т и р (газообразное состояние низкомолекулярного вещества соответствует температуре, существенно выше Ткип, а парообразное – ниже Ткип) основным процессом является сорбция, включающая абсорбцию (растворимость) низкомолекулярного вещества в объеме и его адсорбцию на поверхности полимера. При неразвитой поверхности полимеров и их близком сродстве к низкомолекулярным веществам, что проявляется, в первую очередь, в случае паров органических веществ или воды и близких к ним по полярности полимеров, превалирующую роль играет абсорбция (растворимость) (см. ниже Раздел 2.2).
При развернутой поверхности полимеров и их малом сродстве к низкомолекулярным веществам, в частности, в случае инертных газов и слаборастворимых паров, определяющее значение имеет адсорбция. При этом за разделяющую поверхность принимается поверхность твердой (полимерной) фазы, а величина адсорбции оценивается количеством молекул газа или пара, связанным с этой поверхностью. [5]
При низком давлении газа или пара изменение химического потенциала адсорбируемого вещества (адсорбата или компонента 1):

где 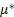 - стандартный химический потенциал;
- стандартный химический потенциал; 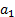 – активность, равная давлению адсорбируемого газа при его низких значениях или, в случае пара, отношению давления пара к давлению насыщения.
– активность, равная давлению адсорбируемого газа при его низких значениях или, в случае пара, отношению давления пара к давлению насыщения.
В данном случае адсорбционное уравнение Гиббса (2.1) может быт записано в виде:
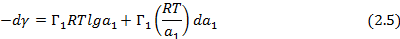
Таким образом, избыток адсорбата (компонента 1) может быть рассчитан по изменению поверхностной энергии при различной активности пара или различном давлении газа.
В соответствие с молекулярно-кинетической теорией адсорбции (теорией Лэнгмюра) адсорбция инертных газов с очень слабым (дисперсионным) межмолекулярным взаимодействием на твердых поверхностях, в том числе полимерных, содержащих равномерно распределенные активные центры со сферой действия, распространяющейся на одну адсорбируемую молекулу, адсорбция рассматривается как процесс установления равновесия адсорбция-десорбция, т.е. равенства скоростей конденсации молекул на активных центрах и их отрыва (испарения) с образованием некоторого равновесного адсорбционного слоя [3].
В изобарно-изотермических условиях кинетика насыщения адсорбционного слоя имеет вид, показанный на рисунке 2.1.1:
| Г(t) |
| Г∞ |
| t |
Рисунок 2.1.1 Схема кинетики образования равновесного насыщенного (мономолекулярного) адсорбционного слоя [5].
где Г(∞) – масса молекул в адсорбционном слое, или равновесная адсорбция.
При заданном давлении адсорбция инертных газов сравнительно слабо зависит от температуры в соответствие с уравнением Клапейрона-Клаузиуса (2.6) из-за малых значений энтальпии (теплового эффекта) адсорбции ΔНа, но сильно зависит от давления при заданной температуре.

В изотермических условиях с увеличением давления газа, в пределе, образуется насыщенный мономолекулярный адсорбционный слой, т.е. достигается максимальная равновесная адсорбция 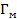 . Схематически вид изотермы адсорбции Лэнгмюра в координатах Г(∞)-р показан на рисунке 2.1.2, кривая 1.
. Схематически вид изотермы адсорбции Лэнгмюра в координатах Г(∞)-р показан на рисунке 2.1.2, кривая 1.
| p |
| Гм |
| Г∞(p) |
| (1) |
| (2) |
Рисунок 2.1.2 Схема изотермы адсорбции Лэнгмюра (1) и БЭТ (2) [5].
Изотерма Лэнгмюра описывается уравнением:

где коэффициент b характеризует отношение констант скоростей процессов десорбции и адсорбции молекул.
Если адсорбция газов на твердой поверхности определяется Ван-дер-ваальсовскими силами, радиус действия которых больше размеров адсорбируемых молекул, то адсорбция не ограничивается мономолекулярным слоем и при высоком давлении приводит к образованию полимолекулярного слоя. Теоретически изотерма полимолекулярной адсорбции, проявляющейся в случае слаборастворимых (инертных) газов и паров на развернутой поверхности полимеров описывается уравнением Брунауэра-Эммета-Тейлора (БЭТ) [5]:
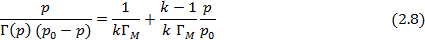
где 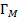 – величина адсорбции при образовании мономолекулярного слоя,
– величина адсорбции при образовании мономолекулярного слоя, 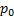 –давление насыщенного пара,
–давление насыщенного пара, 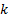 – коэффициент, учитывающий разницу между теплотой адсорбции до образования монослоя и теплотой конденсации при образовании полимолекулярного слоя.
– коэффициент, учитывающий разницу между теплотой адсорбции до образования монослоя и теплотой конденсации при образовании полимолекулярного слоя.
Примерами такой адсорбции является адсорбция слабополярных газов или паров неполярных жидкостей на поверхности полярных полимеров и наоборот, сильнополярных газов или паров полярных жидкостей на поверхности неполярных полимеров,
Такая изотерма имеет так называемый S-образный вид и схематически показана на рисунке 2.1.2, кривая 2. При этом адсорбция при заданном давлении более сильно зависит от температуры по сравнению с Лэнгмюровской вследствие больших значений энтальпии адсорбции.
На практике экспериментальное определение изотерм адсорбции инертных газов или паров на поверхности полимеров используется, главным образом, для определения их удельной поверхности, т.е. площади поверхности, приходящейся на единицу массы полимера, по величине адсорбции в мономолекулярном слое 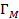 (в молях газа или пара на 1 г полимера):
(в молях газа или пара на 1 г полимера):

где 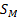 - площадь, занимаемая одной молекулой,
- площадь, занимаемая одной молекулой, 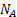 – число Авогадро.
– число Авогадро.
Если изотерма подчиняется уравнению Лэнгмюра, то Гм определяется непосредственно по предельной величине адсорбции, а если уравнению БЭТ – то расчетом по параметрам прямой, полученной перестройкой изотермы в координатах 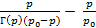 .
.
Равновесная абсорбция
Сорбция (поглощение) низкомолекулярных веществ (газов и паров) полимерами включает в себя адсорбцию на поверхности и объемное поглощение (абсорбцию, или растворимость). Первый процесс является превалирующим при развернутой поверхности полимеров и их малом термодинамическом сродстве с низкомолекулярными веществами, и о нем говорилось в разделе 2.1. Во всех других случаях превалирует абсорбция (растворимость), но ее обычно называют просто сорбцией. При этом поглощаемое вещество называется сорбатом, а поглощающее – сорбентом.
Мерой термодинамического сродства (способности к взаимному растворению, или совместимости) органических полимеров между собой и с другими веществами, в том числе газами, парами и жидкостями в изобарно-изотермических равновесных условиях, служит разность свободных энергий взаимодействующих компонентов в отдельности в двух случаях:
1) в свободном состоянии
2) в молекулярной смеси (растворе) [5]
Это так называемая свободная энергия смешения 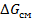 :
:
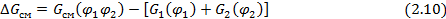
где 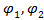 – объемные доли компонентов (1 – низкомолекулярный компонент - жидкость, пар или газ; 2 – полимер);
– объемные доли компонентов (1 – низкомолекулярный компонент - жидкость, пар или газ; 2 – полимер); 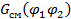 ,
, 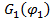 ,
, 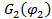 – свободные энергии смеси и отдельных компонентов соответственно.
– свободные энергии смеси и отдельных компонентов соответственно.
При этом определяется энтальпией ( 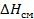 ) и энтропией (
) и энтропией ( 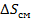 ) смешения:
) смешения:

Условиями термодинамической совместимости (растворимости) компонентов при любом их соотношении при заданных условиях являются: отрицательное значение и вогнутость кривой ее зависимости от 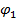 и
и 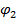 . Определение пределов совместимости используется для получения обобщенных диаграмм фазовых состояний смесей компонентов [5].
. Определение пределов совместимости используется для получения обобщенных диаграмм фазовых состояний смесей компонентов [5].
Для оценки равновесной растворимости одного из компонентов в другом используется разность парциальной мольной свободной энергии (химического потенциала) соответствующего компонента в свободном состоянии и в растворе. В соответствие с теорией Флори-Хаггинса-Скотта для растворов линейных полимеров в низкомолекулярных веществах, изменение химического потенциала низкомолекулярного компонента (газа, пара или жидкости,), растворенного в полимере, по сравнению со свободным состоянием 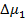 описывается уравнением:
описывается уравнением:
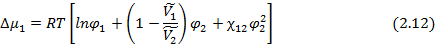
где 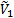 и
и 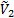 - парциальные мольные объемы низкомолекулярного вещества и полимера соответственно;
- парциальные мольные объемы низкомолекулярного вещества и полимера соответственно; 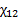 - параметр взаимодействия полимер- низкомолекулярное вещество (параметр Флори-Хаггинса).
- параметр взаимодействия полимер- низкомолекулярное вещество (параметр Флори-Хаггинса).
В этом уравнении два первых члена в скобках соответствуют энтропийному, а третий член – энергетическому (энтальпийному) вкладу вследствие перемешивания молекул компонентов и изменения их межмолекулярного взаимодействия при перемешивании соответственно. Отношение 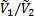 примерно равно степени полимеризации полимера x2 и, следовательно, для высокомолекулярных полимеров и низкомолекулярных веществ обратное отношение
примерно равно степени полимеризации полимера x2 и, следовательно, для высокомолекулярных полимеров и низкомолекулярных веществ обратное отношение 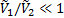 . Параметр Флори-Хаггинса, характеризующий различие в энергии межмолекулярного взаимодействия компонентов в свободном конденсированном состоянии (11, 22) и в растворе (12). В случае идеальных растворов, для которых эти взаимодействия равны,
. Параметр Флори-Хаггинса, характеризующий различие в энергии межмолекулярного взаимодействия компонентов в свободном конденсированном состоянии (11, 22) и в растворе (12). В случае идеальных растворов, для которых эти взаимодействия равны, 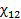 =1/2, а в случае реальных растворов описывается уравнением Гильдебранда[5]:
=1/2, а в случае реальных растворов описывается уравнением Гильдебранда[5]:

где 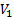 - мольный объем низкомолекулярного вещества,
- мольный объем низкомолекулярного вещества, 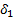 ,
, 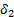 – параметры растворимости компонентов.
– параметры растворимости компонентов.
Для газов и паров изменение химического потенциала:

где а1 – активность, равная для паров отношению давления пара к давлению насыщенного пара, для жидкостей – единице, для газов при сравнительно небольших давлениях – давлению газа.
При этом уравнение Флори-Хаггинса-Скотта (2.12) для линейных полимеров может быть записано в следующем виде:
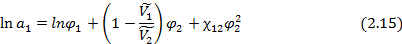
откуда при можно получить выражение для равновесной объемной доли сорбированного низкомолекулярного вещества:

При φ2→1, т.е. если сорбция сравнительно невелика (φ1 << φ2), это выражение упрощается:

Из этих соотношений следует, что чем больше разность параметров растворимости сорбата и полимера, т.е. чем больше их параметр взаимодействия, тем меньше количество равновесно сорбируемого линейным полимером низкомолекулярного вещества. [5]
В случае сетчатых полимеров помимо чисто термодинамических вкладов (энтропийного и энергетического) в изменение свободной энергии и химического потенциала низкомолекулярного вещества при его сорбции существенный вклад вносит упругое сопротивление полимерной сетки увеличению объема полимера при сорбции низкомолекулярного вещества. Учет этого эффекта позволяет записать уравнение Флори-Хаггинса-Скотта ( ) в виде уравнения Флори-Ренера:
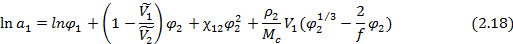
где 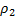 – плотность исходного полимера,
– плотность исходного полимера, 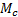 –средняя молекулярная масса полимерных цепей между узами сетки (отношение
–средняя молекулярная масса полимерных цепей между узами сетки (отношение 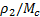 характеризует густоту сетки , или частоту ее узлов и равно числу молей цепей между узлами сетки в единице объема полимера
характеризует густоту сетки , или частоту ее узлов и равно числу молей цепей между узлами сетки в единице объема полимера 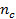 ); f – функциональность полимерной сетки, равная числу цепей, сходящихся в одном узле (чаще всего равная 3 ил 4).
); f – функциональность полимерной сетки, равная числу цепей, сходящихся в одном узле (чаще всего равная 3 ил 4).
Для равновесной объемной доли сорбированного низкомолекулярного вещества при этом получается выражение:
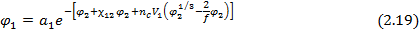
При φ2→1, т.е. если сорбция сравнительно невелика (φ1 << φ2) это выражение упрощается:
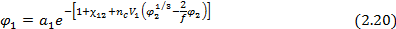
При этом необходимо помнить, что с термодинамической точки зрения жидкое состояние соответствует равенству единице активности ее паров, и в этом случае lna1=0. Однако, очевидно, что сорбция паров при их активности, равной единице, т.е. при р=р∞, и поглощение этого же вещества из жидкой фазы может существенно различаться из-за различия в процессах адсорбции паров на полимерах и смачивания полимеров жидкостями, предшествующих растворению низкомолекулярного вещества, а также процессами растворения (экстракции) низкомолекулярных примесей, содержащихся в полимерах, низкомолекулярной жидкости, что не может происходить в случае насыщенного пара при отсутствие его конденсации на полимере [5].
На практике равновесная сорбция обычно определяется в количестве граммов сорбированного вещества на 1 грамм исходного полимера (в массовой, или весовой концентрации сорбата m1∞, г/г), которая может быть пересчитана в его объемную или мольную концентрацию (с1∞, см3/см3 или С1∞, моль/см3 соответственно), объемные (φ=V1/V1+V2) и весовые, или массовые (g=G1/G1+G2) доли компонентов, где V и G - объем и масса компонентов, через их исходные удельные объемы (v) или плотности (ρ), по следующим соотношениям (для нормальных условий):
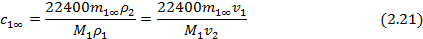

где М1 и V1 – молекулярная масса и мольный объем сорбата соответственно; 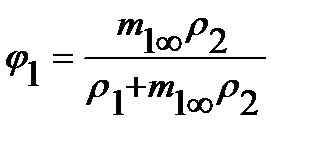 ; φ2=1-φ1;
; φ2=1-φ1; 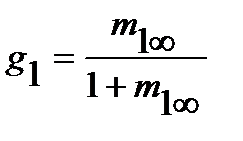 ; g2=1-g1.
; g2=1-g1.
При этом плотность сорбата, поглощаемого из газо- или парообразного состояния, соответствует плотности данного вещества в конденсированном жидком состоянии при заданной температуре [3].
На практике при оценке сорбции низкомолекулярных веществ полимерами, в первую очередь при поглощении жидкостей редкосетчатыми эластичными полимерами, большое значение имеет увеличение объема полимера, или его набухание, степень которого q характеризуется отношением объема полимера с сорбатом к его исходному объему. При аддитивности объемов сорбата и сорбента равновесная степень набухания в первом приближении равна: q= (V1+V2 )/V2, т.е.q=1/ φ2.
Зависимость количества сорбированного вещества от активности пара или давления газа при заданной температуре называется изотермой сорбции, которая в условиях термодинамического равновесия имеет, в соответствие с уравнениями Флори-Хаггинса-Скотта и Флори-Ренера, вид вогнутой кривой (Рисунок 2.2.1, кривая 1).
| m1∞ |
 1 1
|
| 1 |
| 3 |
| 2 |
Рисунок 2.2.1 Изотермы сорбции паров растворителя аморфным полимером в исходном высоко-эластическом или вязко-текучем состоянии (1) и в исходном стеклообразном состоянии при его сохранении в процессе сорбции (2) и переходе в высоко-эластическое состояние с проявлением эффекта “отжига” (3). [5]
| φ кр |
| φ 1 |
| TC |
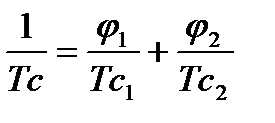 .
.
Рисунок 2.2,2 Зависимости Тс системы полимер-сорбат от объемной доли сорбата: при φ1 < φкр полимер в условиях эксперимента находился в стеклообразном неравновесном состоянии, при φ1 > φкр – в высоко-эластическом равновесном состоянии. [5]
Если такой переход не происходит, т.е. в условиях, когда Тс2 >Тэксп, то полимер находится в неравновесном стеклообразном состоянии на всех стадиях сорбции. При этом условия установившейся сорбции, значительно меньшей по величине равновесной, соответствуют некоторому «квазиравновесному» состоянию полимер-сорбат, и, при отсутствии развернутой поверхности, сорбция протекает по механизму накопления молекул сорбата в «замороженном» свободном объеме полимера с их закреплением по активным центрам на начальных стадиях (при малой активности пара или малом давлении газа) и их агрегированием (кластеризацией) на конечных стадиях (при высокой активности пара или высоком давлении газа). В этом случае изотерма сорбции обычно S-образный вид, соответствующий изотерме адсорбции БЭТ (см. рис.2.1.2 и 2.2.1, кривые 2). [5]
При переходе аморфного, стеклообразного в исходном состоянии, полимера в высокоэластическое состояние в процессе сорбции ее изотерма должна сочетать характер изотермы БЭТ на начальных участках и изотермы Флори-Хаггинса-Скотта или Флори-Ренера – на конечных. При этом пластифицирующий эффект сорбата в полимере может оказывать эффект, аналогичный отжигу полимера и сопровождаться объемной усадкой полимера вследствие уменьшения «замороженного» свободного объема или дезориентацией, если предыстория полимера включала его закалку или эффекты ориентации соответственно. Все это должно резко проявиться на виде изотермы сорбции.
В случае аморфно-кристалличеких полимеров, сорбция паров и газов протекает преимущественно в аморфных областях, являющихся дефектными по сравнению с упорядоченными кристаллическими областями. Величина сорбции газов или паров такими полимерами может быть рассчитана через степень кристалличности (φкр):
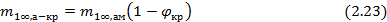
Вид изотермы сорбции будет зависеть от реологического состояния аморфных областей (высокоэластического или стеклообразного) в исходном состоянии и от того, произойдет ли в процессе сорбции их переход из стеклообразного состояния, если они исходно находились в таком состоянии, в высокоэластическое или нет, аналогично некристаллизующимся полимерам. При переходе из стеклообразного состояния в высокоэластическое, помимо эффектов усадки свободного объема и дезориентации в аморфных областях, возможны также эффекты установления более равновесного состояния кристаллических областей, сопровождающихся увеличением степени кристалличности и даже перестройки структуры этих областей. Очевидно, что такие эффекты структурирования должны сказываться на характере изотермы сорбции на предельных стадиях [5].
При сорбции полимерами инертных газов часто наблюдается линейный вид изотермы, т.е проявляется прямая пропорциональность между количеством растворенного газа (в объемной концентрации с1∞,см3/см3) и его давлением (закон Генри): m1∞=σр или с1∞=σр, где σ – константа уравнения Генри, или коэффициент растворимости (сорбции) с размерностью г/г атм или см3/см3атм соответственно. Пересчет σ из одной размерности в другую производится по формуле: σ[см3/см3атм]=22400σ[г/гатм]ρ2/М1ρ1. Из термодинамического анализа растворимости низкомолекулярных веществ полимерами (см. уравнения Флори-Хаггинса-Скотта и Флори-Ренера) в случае равновесной сорбции газов линейными полимерами в высокоэластическом или вязко-текучем аморфном состоянии σ пропорционален еxp[-(1+χ12)]. Зависимость равновесного коэффициента сорбции от температуры описывается уравнением Клапейрона-Клаузиуса через теплоту растворения:

Диффузия воды полимерами
Сорбция и диффузия воды полимерами являются важнейшими характеристиками, в значительной мере определяющими их диэлектрические свойства и свойства на межфазных границах раздела слоев. Полимеры способны впитывать значительное количество воды, что в свою очередь отрицательно сказывается на, собственно, их диэлектрических свойствах. Особенно критично это для тонких полимерных пленок [6].
Диффузия – распространение вещества в какой-либо среде в направлении убывания его концентрации, обусловленная тепловым движением ионов, атомов и молекул. Для одномерной диффузии в идеальных растворах при отсутствии внешних сил справедлив первый закон Фика:

где 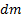 - масса вещества, переносимого при диффузии вдоль оси
- масса вещества, переносимого при диффузии вдоль оси 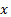 за время
за время 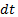 сквозь площадку
сквозь площадку 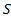 , расположенную перпендикулярно к оси
, расположенную перпендикулярно к оси 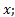
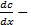 градиент концентрации;
градиент концентрации; 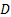 -коэффициент диффузии.
-коэффициент диффузии.
Коэффициент диффузии имеет большое практическое значение, так как в значительной степени определяет скорость многих физико-химических процессов (адсорбции, десорбции и тд) [6].
Явления массопереноса вещества определяют важнейшие свойства полимеров, обусловленные взаимодействием полимера с веществом в объеме материала. Одним из основных механизмов переноса вещества в полимерах является диффузионный.
В зависимости от условий протекания этого процесса различают: взаимодиффузию (наблюдается при наличии градиента концентрации) и самодиффузию. Для описания процесса диффузии применяются два подходи: феноменологический и молекулярно-кинетический. Феноменологический подход основан на первом законе Фика. Поскольку поток вещества стремится устранить неравномерность распределения вещества в системе, то коэффициент диффузии является мерой скорости, с которой система способна при заданных условиях выравнивать разность с концентрацией. Эта скорость в свою очередь связана с микроскопическими параметрами системы, характеризующих тепловую подвижность макромолекул диффузионной среды и диффундирующих частиц температурой, структурой полимера и природой диффундирующего вещества[6].
Второе уравнение Фика, характеризующее нестационарный процесс диффузии, выражает изменение концентрации вещества в различных точках пространства как функцию времени:

Если диффузия протекает в трех направлениях изотропной среды:

В случае анизотропной среды перенос вещества в нестационарном состоянии описывается уравнением:
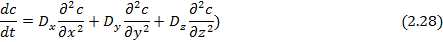
Для расчета диффузии необходимо решить уравнения с определенными граничными условиями и начальными условиями, которые в свою очередь задаются параметрами исследуемого процесса.
При наличии макроскопических дефектов - трещин, пор, отверстий – в твердом теле мы имеем дело не с диффузионным межмолекулярным переносом, а с фазовым переносом, описываемым законом Пуазейля для газов и паров[5]. Деффекты рассматриваются в таком случае как трубки и капилляры. Как правило, данные дефекты наиболее ярко выражены в тонких пленках, и их наличие носит критический характер. Закон Пуазейля применяется в случае, когда длина пробега диффундирующего вещества меньше длины трубки. Тогда массоперенос можно оценить по закону Пуазейля:
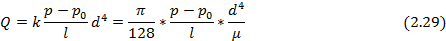
где 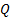 - объем газа, прошедшего за 1 секунду через сечение трубки;
- объем газа, прошедшего за 1 секунду через сечение трубки; 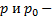 давление в двух сечениях трубки;
давление в двух сечениях трубки; 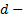 диаметр трубки,
диаметр трубки, 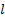 - расстояние между сечениями;
- расстояние между сечениями;  - коэффициент вязкости.
- коэффициент вязкости.
В случае фазового переноса коэффициент диффузии имеет значения до 10-3 до 10-1 см2/с, в то время как коэффициент диффузии, описываемый законом Фика, имеет гораздо более низкие значения: от 10-12 до 10-6 см2/с. Таким образом, зная порядок значения коэффициента диффузии в системе, можно сделать вывод о характере процесса массопереноса.
Молекулярно-кинетический подход рассматривает диффузию как последовательность элементарных трансляционных движений молекул диффузанта в полимерной матрице. (т.н. диффузионный скачок) [6].
Заключение
Установлено, что к диэлектрическим свойствам относятся: диэлектрическая проницаемость, диэлектрические потери и тангенс угла диэлектрических потерь. На их величину основное влияние оказывает полярность молекул, которая, в свою очередь, определяется химическим составом полимерных молекул и температура, влияющая на время релаксации соответствующих форм тепловой подвижности. Показано, что на диэлектрические свойства полимера существенное влияние оказывает наличие в его составе низкомолекулярных веществ, которые приводят к проявлению пластифицирующего эффекта, что приводит к изменениям свойств, соответствующим повышению температуры.
Показано, что сорбция делится на абсорбцию и адсорбцию, а последняя в свою очередь на моно- и полимолекулярную. При развитой поверхности полимера в изобарно-изотермических условиях кинетика насыщения адсорбционного слоя слабо зависит от температуры , но сильно зависит от давления, и описывается уравнением изотермы Лэнгмюра. Если адсорбция газов на твердой поверхности определяется Ван-Дер-Ваальсовскими силами, то образуется полимолекулярный слой и адсорбция описывается уравнением БЭТ. Выведено, что при неразвитой поверхности полимеров и их близком сродстве к НМВ, превалирующую роль играет абсорбция. Мерой термодинамического сродства является свободная энергия смешения. Для оценки равновесной растворимости одного из компонентов в другом используется химический потенциал. Диффузия воды влияет на диэлектрические свойства и определяет скорость многих физико-химических процессов. Описывается законами Фика, а при наличии макроскопических дефектов - законом Пуазейля.
Рассмотрено поведение тонких пленок на основе полиамида, полиимида, поливинилового спирта и отвержденной алифатическим амином смолы ЭД-20. Сорбция паров воды в ПА сопровождается значительным (в 1000 раз) возрастанием электрической. Кинетика изменения емкости структуры на частоте 1 МГц значительно выше, чем на более низких частотах. Наблюдается прямая пропорциональность между концентрацией воды и изменением емкости МДД при S-образной форме изотерм сорбции, что свидетельствует о роли как количества, так и состояния сорбированной воды на изменение емкости МДД. Состояние сорбированной воды зависит от величины адсорбции , и связано с ростом и поведением кластеров воды на адсорбционных центрах.
Список литературы
1. Перепечко И.И. «Введение в физику полимеров» «Химия», 1978 г.
2. Н.А.Козлов, А.Д.Митрофанов «Физика полимеров» Владимирский государственный университет, 2001 г.
3. Б.И. Сажин, А.М. Лобанов, и др. «Электрические свойства полимеров» / Под редакцией Б.И. Сажина – 3-е изд. перераб. – Л.: Химия, 1986 г.
4. В. Ф. Важов, В. А. Лавринович «Техника высоких напряжений» Курс лекций, Издательство ТПУ,Томск, 2008 г.
5. П.Г. Бабаевский Курс лекций «Физика полимеров» 2018 г.
6. П.Г. Бабаевский, Н.А. Козлов , А.А. Жуков, С.А Жукова «Определение кинетики сорбции воды и коэффициентов диффузии в тонких полимерных покрытиях емкостным методом с помощью микродиэлектрических датчиков» Методическое пособие, 2004 г.
7. Е.А. Тутов, Е.Н. Бормонтов и др. «МДП структура с полиамидным диэлектриком в условиях сорбции паров воды» Журнал технической физики, том 75, вып. 8, 2005 г.
8. П.Г. Бабаевский, Н.А. Козлов , А.А. Жуков «Исследование сорбции и диффузии воды в полимерных покрытиях диэлектрическим методом» 2001 г.
9. И. Н Муллагалиев, И. Н. Сафаргалин, Р. Б. Салихов «Химические датчики на основе тонких полимерных пленок» Электротехнические и информационные комплексы и системы. № 3, т. 13, 2017 г.
10. В.А. Белоглазов «Дипольный момент молекул и диэлектрическая проницаемость адсорбированной воды в полиимидной пленке ПМ» 2000 г.
Институт материаловедения и технологий материалов
Дата: 2019-07-31, просмотров: 420.