Калужский Государственный Педагогический
Университет им. К.Э. Циолковского
Кафедра философии и социологии
Электроосаждение металлов.
Калуга, 2008г.
Содержание
Глава 1 История
1.1. СТРАНИЦЫ ИСТОРИИ
1.2. СТРУКТУРА ЭЛЕКТРООСАЖДЕННЫХ МЕТАЛЛОВ
1.3. ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ – ОСНОВА ЭЛЕКТРОХИМИЧЕСКИХ ТЕХНОЛОГИЙ
Глава 2. Электрохимическое выделение металлов
2.1. ОБЩАЯ ХАРАКТЕРИСТИКА ПРОЦЕССОВ
2.2. ОБРАЗОВАНИЕ МОНОАТОМНЫХ СЛОЕВ МЕТАЛЛОВ ПРИ ПОТЕНЦИАЛАХ ПОЛОЖИТЕЛЬНЕЕ РАВНОВЕСНЫХ
2.3. ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ПРОЦЕССЫ КАТОДНОГО ВЫДЕЛЕНИЯ МЕТАЛЛОВ
2.3.1. Роль природы металла
2.3.2. Роль состава раствора
2.4. ПРИРОДА МЕТАЛЛИЧЕСКОГО ПЕРЕНАПРЯЖЕНИЯ
2.5. ФАКТОРЫ, ОПРЕДЕЛЯЮЩИЕ ВЕЛИЧИНУ ПОЛЯРИЗАЦИИ ПРИ КАТОДНОМ ВЫДЕЛЕНИИ РАЗЛИЧНЫХ МЕТАЛЛОВ
2.5.1. Энергия иона в металле и его состояние в растворе
2.5.2. Активность поверхности катода в процессе осаждения металлов
2.5.3. Заряд поверхности металла в условиях его катодного осаждения
2.5.4. Другие возможные причины появления металлического перенапряжения
Заключение
ГЛАВА 1 ИСТОРИЯ
СТРАНИЦЫ ИСТОРИИ
Электрохимия принадлежит к числу тех немногих наук, дата рождения которых может быть установлена с высокой точностью. Это рубеж XVIII и XIX веков, когда благодаря знаменитым опытам итальянского физиолога Л. Гальвани и созданию итальянским физиком А.Вольта в 1799 году «вольтова столба» — первого в истории человечества химического источника тока — были сформулированы проблемы, решение которых определило основные задачи электрохимии.
«Без химии путь к познанию истинной природы электричества закрыт» — эта мысль принадлежит великому М.В.Ломоносову, который сделал исключительно важные наблюдения и замечания о природе пассивности металлов. Как под влиянием Ломоносова, так и в соответствии с общей ситуацией в науке, в Московском университете электрохимические исследования всегда занимали важное место и при этом оказывались чрезвычайно результативными [1–3]. Так, в 1807 г. российский академик Ф.Ф.Рейсс открыл явления электроосмоса и электрофореза. Значительно опередили свое время работы профессора физики А.П.Соколова, впервые применившего переменный ток для определения емкости электрода (1887 г.).
Фундаментальный вклад в теорию растворов электролитов внес профессор И.А.Каблуков [4], указавший пути к объединению гидратной теории растворов Д.И.Менделеева и теории электролитической диссоциации С.Аррениуса на основе представления о диссоциации как взаимодействии с растворителем. Каблуков исследовал свойства неводных растворов и открыл явление аномальной электропроводности. Как автор известного учебника по физической химии и электрохимии (первое издание — 1902 г.) И.А.Каблуков оказал огромное влияние на распространение электрохимических знаний в нашей стране. Он был твердо уверен в том, что «без знания электрохимии нельзя приступить к решению многих вопросов не только в области минеральной, но и органической химии; на законах электрохимии основаны методы исследования различных вопросов химической механики...».
В 1911 г. Е.И. Шпитальским было открыто явление электрополировки металлов, которое изучалось в дальнейшем сотрудниками и студентами лаборатории физической химии под его руководством. Одновременно проводились работы и по другим вопросам теоретической и технической химии (электрохимическое получение бертолетовой соли, исследования хлорного электролиза, разработка новых электролитических способов получения свинцового глета и окиси меди и т.д.). В этих работах участвовали Н.Н.Петин, З.А.Иофа, А.В.Командин, В.В.Монбланова и др. Е.И.Шпитальский организовал также исследования реакций выделения водорода и кислорода на платиновых электродах с помощью специально разработанного коммутаторного метода. Изучалось влияние предварительной анодной и катодной обработки платины и палладия на их каталитическую активность в реакции разложения перекиси водорода (М.Я.Каган).
В 1929 г. Е.И. Шпитальский был избран членом-корреспондентом Академии наук СССР и в том же году его научная деятельность оборвалась: он был осужден по делу Промпартии.
Из более поздних работ, проводившихся уже после создания в 1929 году кафедры физической химии, особо следует отметить исследование Н.И.Кобозева и Н.И.Некрасова по механизму электрохимического выделения водорода на металлах (1930 г.). В этой работе впервые было указано на роль энергии связи адсорбированных атомов водорода с металлом в этом процессе. Н.И.Кобозев и В.В.Монбланова впервые использовали электрохимическую ячейку, разделенную палладиевой мембраной, для исследования диффузии атомарного водорода через палладий.
Анализируя влияние природы металла на скорость электрохимического выделения водорода, Н.И.Кобозев и В.В.Монбланова ввели понятие об электрокатализе как явлении ускорения электродной реакции в результате адсорбционного взаимодействия реагента или интермедиата с металлом электрода.
Таким образом, к началу 30х годов на кафедре физической химии работы по электрохимии проводились достаточно интенсивно, что привело к появлению специализации по электрохимии.
В 1930 г. лабораторию технической электрохимии МГУ возглавил А.Н.Фрумкин (1895–1976) — автор фундаментальных экспериментальных и теоретических работ по равновесным свойствам заряженных межфазных границ, блестяще образованный ученый мирового класса, работы которого, несмотря на молодость автора, уже стояли в одном ряду с трудами Нернста, Липпмана, Лэнгмюра. В 1932 г. он был избран действительным членом Академии наук СССР. Под руководством академика Фрумкина лаборатория технической электрохимии была преобразована в 1933 году в кафедру электрохимии. В этот период ближайшими сотрудниками А.Н.Фрумкина в МГУ были З.А.Иофа и А.И. Шлыгин, которые работали до этого в области электрохимии как сотрудники кафедры физической химии. А.Н.Фрумкин руководил кафедрой до 1976 г.; с 1976 по 1998 гг. кафедру возглавлял проф. Б.Б.Дамаскин, а с ноября 1998 г. кафедрой заведует проф. О.А.Петрий.
Общие понятия
Технология – наука о ремеслах, о производстве. Электрохимические технологии (ЭХТ) многообразны и могут быть для облегчения ознакомления с ними разбиты на ряд разделов в соответствии с выбранным классификационным признаком. Например, в соответствии с природой производимых продуктов или услуг. По мере развития электрохимических технологий изменялись содержание и масштабы использования ее разделов. В основе всех электрохимических технологий лежат электрохимические процессы в электрохимических системах на границах электрод-электролит (межфазных границах). Они характеризуются, как и другие химические процессы термодинамическими и кинетическими параметрами.
Современный этап развития электрохимии межфазных границ как науки характеризуется, как отмечают специалисты1 как переход от феноменологических к молекулярным подходам. При этом детализация феноменологических подходов играет важную роль в «состыковке» расчетов и теоретических прогнозов молекулярного уровня с экспериментом. Выделяют следующие центральные проблемы:
- Разработка молекулярных моделей строения межфазных границ и адсорбционных слоев.
- Теоретическое описание и молекулярное моделирование элементарных стадий переноса заряженных частиц через межфазные границы.
- Установление кинетики и механизмов электрокаталитических процессов и создание моделей, учитывающих молекулярное строение адсорбционных слоев.
- Развитие феноменологических и микроскопических представлений о механизмах образования и роста новых фаз и коррозионно-электрохимических явлений.
- Создание теоретических основ электрохимических методов получения материалов с заданными (нано)структурой и свойствами и определение принципов функционирования таких материалов. Выявление специфических особенностей электрохимических явлений в наноразмерных системах.
- Совершенствование представлений о сложных стадийных электродных процессах на основе прямого наблюдения их интермедиатов и новых решений макрокинетических задач.
Большинство сформулированных выше фундаментальных проблем существовало в электрохимии межфазных границ на протяжении последних 30-50 лет, и современный этап характеризуется лишь смещением некоторых акцентов. В то же время формулировкой наноэлектрохимических проблем и задач наука полностью обязана прогрессу последнего десятилетия.
Достижения электрохимической науки определяют современное состояние и направления совершенствования ЭХТ.
Электрохимические технологии – это технологические процессы, используемые в производстве товаров и услуг, в основе которых лежат электрохимические процессы.
К основным группам ЭХТ относятся:
· электрохимическая энергетика;
· электрохимическая металлургия;
· электрохимические технологии химических товарных продуктов;
· электрохимические технологии в производстве изделий и инструмента в машиностроении и приборостроении;
· электрохимические технологии, используемые для защиты от коррозии объектов техники;
· электрохимическая сенсорика;
· электрохимические технологии в здравоохранении;
· электрохимические технологии пищевой воды.
Некоторые из классов современных электрохимических технологий в наиболее общих чертах рассмотрены.
Сильные и слабые стороны электрохимических технологий
Преимущества.
1. При производстве химических продуктов с помощью электрохимической технологии достигается высокая селективность электрохимических процессов, так как процессы окисления и восстановления на электродах протекают с участием электронов (а не сложных химических соединений в качестве реагентов).
2. В электрохимических процессах легко изменяется потенциал изменением тока, протекающего через электрод, достигая таких значений окислительно-восстановительного потенциала, которые создают уникальные возможности для процессов окисления и восстановления.
3. С использованием диафрагм или ионообменных мембран в электролизерах оказывается возможным одновременно получать несколько чистых продуктов (например, при электролизе раствора NaCl получают Cl2, H2 и щелочь).
Недостатки.
1. Большой расход электроэнергии, которая затрачивается на электрохимические превращения.
2.Относительно невысокая производительность электрохимических процессов (гетерогенных процессов, протекающих на границе электрод-раствор). В результате имеют место относительно высокие капитальные затраты на оборудование, здания. В настоящее время нашли широкое применение пористые электроды, позволяющие значительно увеличить удельные поверхности, на которых осуществляются электрохимические реакции. Это позволяет повысить производительность электрохимических устройств за счет повышения плотности тока, в общем случае, путем повышения «амперной нагрузки» на единицу производственной площади. При общей оценке вклада в экономику ЭХТ следует отметить, что только стоимость хлора и щелочи, производимых электролизом, составляет около 10 % общей стоимости продукции химической промышленности. Общая мощность ХИТов, находящихся одновременно в эксплуатации, превышает мощность всех электростанций мира. С экологической точки зрения электрохимические производства характеризуются весьма токсичными продуктами (Cl2 и F2) и использованием в производстве сильно токсичных веществ: ртуть в качестве электрода, асбест в качестве диафрагменного материала и т.д. Поэтому вопросы охраны труда и техника безопасности очень важны! Электрохимический объект - это основной элемент электрохимической технологии. Объект может принадлежать к классу искусственных или естественных электрохимических объектов. Могут быть объекты, являющиеся промежуточными. Искусственные объекты – это электрохимические устройства, естественные – коррозионные процессы. К промежуточным можно отнести безэлектролизные объекты (функционирующие без внешнего тока), в которых проходящие на поверхности технических объектов самопроизвольные окислительно-восстановительные процессы. Сюда относят процессы химической металлизации, травления, оксидирования, фосфатирования, хроматирования,
пассивирования, активирования металлов и другие подобные технологические приемы. Они протекают самопроизвольно без внешнего тока, но условия оптимального протекания создаются искусственно.
В некоторых случаях герметичность ЭХУ нужна для того, чтобы электролиты не взаимодействовали с внешней средой. С помощью штуцеров 13 и 14 устройство может включаться в гидравлическую схему (постоянно или временно)
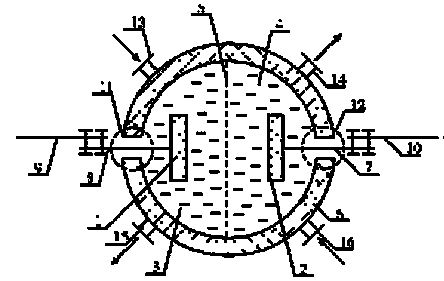 |
Рис. 1.1- Обобщенная схема электрохимического устройства-1, 2 – электроды (анод, катод); 3, 4 – электролиты, находящиеся в анодном и катодном пространствах (анод – анолит, катод - католит); 5 – разделитель (сепаратор, диафрагма, мембрана), предназначен для формирования анодного и катодного пространств; 6 – корпус; 7, 8 –токоотводы от электродов (электронные проводники); 9, 10 –токоотводы от внешней цепи (элементы внешней электрической цепи); 11, 12 – узлы герметичного вывода токоотводов из корпуса электрохимического устройства.
Электроды ЭХУ могут быть твердыми, но могут быть и жидкими, например, таким является расплавленный катод в электролизере для получения алюминия или ртутный катод в хлорном электролизере. По геометрической характеристике они могут быть гладкими или пористыми. Поверхности электродов могут быть покрыты каталитическими слоями, природа которых может быть разнообразна, и определяется спецификой реакций на электроде.
Электроды могут быть газовыделяющими или газопоглощающими (газодиффузионными). Пористая структура электродов обеспечивает увеличение реакционной поверхности и увеличение интенсивности процесса в электрохимических устройствах. Важное место в электрохимических технологиях принадлежит электродам, при формировании которых используются явления гидридообразования и интеркаляции, а также электродам с полупроводниковыми, алмазными, каталитическими (в том числе, биокаталитическими) слоями.
Является в форме электрической энергии, а во втором — в форме теплоты. Для этого возьмем какое-либо химическое превращение, например Cu2+
Если эта реакция протекает как химический процесс, то она будет характеризоваться рядом особенностей.
Реакция возможна только при столкновении ее участников друг с другом. Следовательно, необходимость контакта реагирующих частиц является первой характерной особенностью химического процесса.
В момент столкновения, когда реагирующие частицы вплотную подходят друг к другу, становится возможным переход электронов от одной частицы к другой. Совершится ли такой переход в действительности, зависит от энергии реагирующих частиц и ее соотношения с энергией активации; энергия активации является функцией природы химической реакции и для ионных реакций она обычно невелика. Путь электрона при таком переходе очень мал, что является второй характерной особенностью химического процесса.
Столкновения могут происходить в любых точках реакционного объема и при любых взаимных положениях реагирующих частиц в пространстве, поэтому электронные переходы могут совершаться в любых направлениях в пространстве (рис. 1). Хаотичность, беспорядочность столкновений между реагирующими частицами и пе-направленность электронных переходов являются третьей характерной особенностью химического процесса.
В результате этих особенностей энергетические эффекты химических процессов проявляются в форме теплоты. Чтобы энергетические изменения, соответствующие химическому превращению, проявлялись в виде электрической энергии, т. е. чтобы происходил электрохимический процесс, необходимо изменить условия его протекания.
Получение или затрата электрической энергии всегда связаны с прохождением электрического тока, представляющего собой поток электронов, перемещающихся по одному и тому же пути. Условия протекания химической реакции необходимо поэтому изменить так, чтобы электронные переходы были не беспорядочны, а совершались в одном определенном направлении. Использование энергии электрического тока возможно лишь в том случае, если путь электронов велик по сравнению с размерами атомов. Таким образом, в электрохимических процессах переход электронов от одного участника реакции к другому должен совершаться по достаточно длинному пути. Однако путь электрона не может быть большим, если реагирующие частицы контактируют друг с другом. Поэтому для электрохимического процесса обязательно пространственное разделение участников реакции. Но одного только пространственного разделения недостаточно, так как оно приведет к прекращению химической реакции, а не к превращению ее в электрохимическую. Для осуществления электрохимического процесса необходимы дополнительные условия: электроны должны отрываться от ионов меди и по одному общему пути переходить к ионам железа. Этого можно достичь, заменив непосредственный контакт между участниками реакции их контактом с двумя металлическими телами, соединенными между собой металлическим проводником. Для того чтобы поток электронов был непрерывным, необходимо обеспечить прохождение электрического тока также и через реакционное пространство. Оно обычно осуществляется и участниками электрохимической реакции (если они находятся в ионизированном состоянии), и специально добавленными соединениями, обладающими в данных условиях высокой ионной проводимостью.
При электрохимической реакции прямой контакт между реагирующими частицами заменяется их контактом с соответствующим металлом. При этом реакция и связанные с ней энергетические изменения остаются теми же (независимо от того, протекает она по химическому или же электрохимическому пути), но кинетические условия могут быть различными. Энергия активации при электрохимическом механизме благодаря каталитическим свойствам металлов может быть иной, чем при гомогенном химическом механизме, кроме того, она зависит от потенциала. В электрохимических реакциях обязательно участвуют электроны, а часто и другие заряженные частицы — катионы и анионы, что составляет одну из их основных характерных особенностей. Энергия таких частиц, естественно, является функцией электрического поля, создаваемого на границе электронопроводящее тело — электролит.
Отсюда следует, что и скорость электрохимической реакции зависит не только от температуры, активностей ее участников и катализатора, т. е. от тех же факторов, которые определяют скорость химической реакции, но и от потенциала на границе раздела фаз различной проводимости. Варьирование потенциала границы, при сохранении постоянными концентраций участников электрохимической реакции и температуры, позволяет в десятки, сотни и тысячи раз менять скорость реакции, а в ряде случаев и природу ее продуктов. Это делает электрохимические реакции более управляемыми, легче контролируемыми, чем химические. Электрохимические реакции можно определить как такие химические реакции, скорость которых является функцией потенциала . Поэтому электрохимические реакции отличаются от химических не только по энергетическому эффекту процесса, но также и по величине энергии активации.
Взаимное превращение химической и электрической форм энергии совершается только в электрохимических системах, поэтому их изучение составляет предмет электрохимии.
Электрохимическая система содержит следующие составные части (рис. 2).
|
|
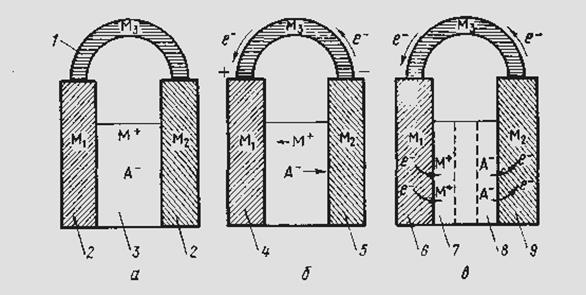
Рис. 2. Схематическое изображение электрохимической системы:
а — равновесная электрохимическая система; б—химический источник тока; в — электрохимическая ванна; / — внешняя цепь; 2 — электроды; 3 — электролит; 4 — положительный электрод; 5 — отрицательный электрод; 6 — катод; 7 — като-лит; 8 — анолит; 9 — анод
1. Реагенты, а также ионизированные или способствующие ионизации реагентов вещества, обеспечивающие прохождение электрического тока; эта часть системы является ионным проводником электричества (проводник II рода) и называется электролитом.
2. Два электронопроводящих тела, контактирующие с электролитом и обеспечивающие обмен зарядами с участниками электрохимической реакции, а также передачу электронов во внешнюю цепь (см. ниже) или получение их из внешней цепи; они называются электродами. На электродах — на границе раздела двух различно проводящих фаз — происходит перенос заряда, т. е. протекают электрохимические реакции, иными словами, именно здесь локализовано взаимное превращение химической и электрической форм энергии. Электроды поэтому следует рассматривать как наиболее важную часть электрохимической системы
3. Металлический проводник (проводник I рода), соединяющий электроды и обеспечивающий прохождение тока между ними; он называется внешней цепью.
Если электролит представляет собой токопроводящий раствор одного или нескольких веществ в воде или ином растворителе, то такие системы относятся к электрохимии водных или неводных растворов; если электролитом служит расплавленная соль (или смесь расплавленных солей и оксидов), эти системы относятся к электрохимии расплавов или расплавленных сред; если межэлектродное пространство заполнено газом — к электрохимии газов. Электрохимическая система может находиться в равновесном (рис. 2, а) или неравновесном (рис. 2, б, в) состоянии.
Электрохимическая система, производящая электрическую энергию за счет протекающих в ней химических превращений, называется химическим источником тока или гальваническим элементом (рис. 2, б). Здесь электрод, посылающий электроны во внешнюю цепь, называется отрицательным электродом или отрицательным полюсом элемента. Электрод, принимающий электроны из внешней цепи, называется положительным электродом или положительным полюсом элемента.
Электрохимическая система, в которой за счет внешней электрической энергии совершаются химические превращения, называется электролизером или электролитической ванной (рис. 2, в). Электрод, принимающий электроны от участников реакции, называется анодом. Электрод, отдающий электроны участникам реакции, — катодом. Часть электролита, примыкающая к аноду, называется анолитом; примыкающая к катоду — католитом.
Поскольку потеря электронов отвечает реакции окисления, а их приобретение — реакции восстановления, то можно сказать, что анод — это электрод, на котором происходит окисление, а катод — электрод, на котором происходит восстановление. Поэтому анод одновременно является отрицательным, а катод — положительным полюсом химического источника тока.
Изложенные соображения о различии электрохимических и химических реакций и о предмете и содержании электрохимии отвечают воззрениям, сложившимся в отечественной литературе. В согласии с расширенным определением электрохимии к ней можно отнести явления, связанные с электрохимическими свойствами коллоидов, с химическими реакциями, вызванными действием света или потока радиоактивных частиц (и приводящими к возникновению разности потенциалов), с электрохимическими явлениями в животных и растительных организмах и т. п. Представляется, однако, более правильным говорить в этих случаях о коллоидной электрохимии, фотоэлектрохимии, радиоэлектрохимии, биоэлектрохимии и т. д., сохранив название собственно электрохимии для
До сих пор рассматривалась роль, которую адсорбция играет лишь непосредственно в самом процессе электролитического восстановления (или окисления). Этот фактор должен сказываться на кинетике конкурирующих процессов, т. е. на кинетике электрохимического выделения водорода (или кислорода). Присутствие адсорбированных веществ на поверхности электрода может и увеличивать и уменьшать перенапряжение водорода (или кислорода). Это, в свою очередь, будет изменять условия протекания реакции восстановления (или окисления). Такого рода представления согласуются с тем экспериментальным фактом, что реакции электроокисления или электровосстановления многих веществ часто протекают со значительным выходом по току при потенциалах, заметно больших, чем те, при которых (при той же плотности тока) происходит выделение водорода или ирииелодит выделение водорода или кислорода из растворов, не содержащих «деполяризатора». В частности, проведение синтеза Кольбе возможно именно потому, что органические соединения, адсорбируясь на платиновом электроде, отравляют его, затрудняя тем самым выделение кислорода и смещая потенциал до значения, при котором уже может начинаться окисление анионов карбоновых кислот. Адсорбция органических веществ при столь положительных и удаленных от нулевой точки металла потенциалах кажется невозможной, если не учитывать окисления поверхности. Нулевая точка окисленного металла, как это было отмечено на примере свинца, сдвинута далеко в положительную сторону по сравнению с ее значением для чистого металла.
ОБРАЗОВАНИЕ МОНОАТОМНЫХ СЛОЕВ МЕТАЛЛОВ ПРИ ПОТЕНЦИАЛАХ ПОЛОЖИТЕЛЬНЕЕ РАВНОВЕСНЫХ
К настоящему времени можно считать доказанным, что на подложке из металла Mi, отличного по своей природе от осаждаемого металла М2, в очень многих случаях процесс осаждения начинается с образования моноатомного слоя, а возникновение и развитие кристаллических зародышей совершается уже на этом слое. Осаждение первого монослоя металла на чужеродной подложке наблюдается при потенциале более положительном, чем равновесный потенциал выделяющегося металла в данном растворе, т. е. при данной активности его ионов. В связи с этим в зарубежной литературе широко используется термин «допотенциальное осаждение» (underpotential deposition), который при буквальном его переводе на русский не передает сущности явления, вместо него поэтому используются термины «осаждение при недонапряжении» или «дофазовое осаждение». По-видимому, первым, кто наблюдал эффект дофазового осаждения металлов, был Гевеши, работы которого относятся к 1912 г. Они были затем основательно забыты, и интерес к этой проблеме возродился лишь в 60—70-х годах, и сейчас она оказалась в центре внимания и электрохимиков и физиков. Очень интересные результаты были получены с применением циклической вольтамперометрии и тонкослойных электрохимических систем. На рис. 22.4 приведена циклическая /—£-кривая, описывающая осаждение таллия на серебряном электроде. Катодный пик тока наблюдается при потенциале примерно на 0,4 В положительнее равновесного потенциала таллия в растворе выбранного состава и отвечает образованию первого монослоя. Второй подъем тока приходится на равновесный потенциал таллия и соответствует выделению компактного осадка таллия, т. е. появлению в системе новой твердой фазы — металлического таллия. Переход в анодную область позволяет наблюдать максимум тока при равновесном потенциале таллия и другой максимум, отвечающий растворению дофазового слоя. Количество таллия, осевшего в области дофазового осаждения, можно с достаточной точностью рассчитать по количеству электричества, потраченного на его растворение при потенциале первого пика. Расчеты показывают, что при дофазовом осаждении образуются обычно полные или незавершенные моноатомные металлические слои. Так как потенциал дофазового осаждения (потенциал пика) ё>П положительнее соответствующего равновесного потенциала &р, то энергия связи атомов первого монослоя с чужеродной подложкой должна быть больше, чем атомов осаждаемого металла с одноименной подложкой. Сдвиг потенциала Дс?п в положительную сторону является следствием повышенной энергии связи A<?m,2 или, с некоторым приближением, повышенной теплоты адсорбции:
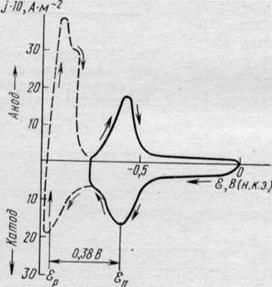
| Рис. 22.4. Циклическая вольтампер иая кривая, полученная на Ag-электроде в 2-Ю-4 М T1NO3 на фоне 0,5 М Na2SO,i; скорость развертки 20-Ю-3 В-с-1 |
Потенциал дофазового осаждения можно описать формулой Нерн-ста для электродов первого рода:
Если принять, что активность металла аи в растущем моноатомном слое отлична от единицы (как это обычно принимается для компактного металла) и зависит от степени покрытия в подложки монослоем,
Поскольку 02f2<l, потенциал монослоя в согласии с опытом оказывается положительнее равновесного потенциала того же металла в том же растворе. Из (22.5) также в согласии с опытом следует, что &П меняется с активностью ионов металла в растворе по такому же закону, как и равновесный потенциал электрода первого рода:
Соблюдение зависимости (22.6) указывает на то, что величина п отвечает заряду иона z , т. е. в образовании монослоя участвуют не ионы или частично разрядившиеся ионы, а атомы металла.
Уравнение (22.5), таким образом, удовлетворительно описывает ряд основных особенностей дофазового осаждения металлов, но не раскрывает его механизм. В этом отношении более перспективными представляются работы, в которых Д£„ сопоставляется с элект-роотрицательностями металлов, работами выхода электронов из них и т. д. По Кольбу, Прзасницкому и Геришеру, между Д£п и разностью работ выхода электрона a«'£ из металла подложки (Mi) и из осаждающегося металла (М2) существует прямо пропорциональная зависимость с наклоном, равным 0,5:
F \ g ]{ = 0,5 Ды1'! . (22.7)
Подобная же зависимость была найдена Трасатти, ко с наклоном, равным единице:
F \ё „ = Wgl . (22.8)
Рассмотрение кривых Д£ —Да>£- с привлечением всех наиболее надежных данных по потенциалам дофазового осаждения и работам выхода показало, что коэффициент пропорциональности k лежит между 0,5 и 1, т. е.
F \(о и — k Дсо ' . /99 Q\
Независимо от величины k из уравнений (22.7) —(22.9) следует, что дофазовое осаждение металлов наблюдается только в том случае, когда работа выхода электрона из металла подложки (металл Mi) больше, чем из металла монослоя (М2). Следовательно, образование монослоя сопровождается переносом электронов из него в субстрат и появлением диполей на границе раздела М, и М2, причем положительный конец диполя расположен на монослое. Свойства монослоя, его структура, во многом определяемая структурой субстрата, играют очень важную роль в процессе дальнейшего развития осадка, влияя также на адсорбционные, каталитические, коррозионные и другие характеристики металла. Дофазовое осаждение представляет поэтому не меньший интерес, чем. зароды-шеобразование, и с ним необходимо считаться при рассмотрении механизма возникновения новой металлической фазы.
Несмотря на все особенности протекания процессов электрохимического выделения металлов, создающие серьезные трудности при проведении экспериментов и при теоретической интерпретации их результатов, к настоящему времени уже накоплен значительный фактический материал и сформировались определенные взгляды на природу этих процессов. Получение достаточно достоверных опытных данных сделалось возможным благодаря развитию техники эксперимента (применение новых методов исследования, при помощи которых удается избежать осложнений, связанных с особенностями построения кристаллической решетки и изменением
потенциала во времени), разработке методики измерения поверхностен роста и, соответственно, истинной плотности тока, тщательной очистке растворов и т. п.
Роль природы металла
При электролизе растворов простых солей характер катодных осадков и величина электродной поляризации определяются в первую очередь природой выделяющегося металла (табл. 22.1).
Таблица 22.1. Классификация металлов по значению металлического перенапряжения при их выделении из растворов простых солей
| группы | Металлы | Перенапряжение, В | 2 Ток обмена, А-м | Средние размеры зерен осадка, м |
| I | Hg, Ag, Tl, Pb, Cd, Sn | 0—П-1Э-3 | п-103- п-13 | >10-5 |
| II | Bi, Cu, Zn | п -13-2 | /;. 13—1!. 13-1 | 10-5—Ю-6 |
| III | Co, Fe, Ni | п-13-1 | п-13-4—п-13-5 | <10-7 |
Все металлы, приведенные в табл. 22.1, можно разделить на три группы. К первой из них относятся металлы, выделяющиеся из водных растворов или совсем без перенапряжения (ртуть), или с очень малым перенапряжением, не превышающим при обычных плотностях тока тысячных долей вольта (серебро, таллий, свинец, кадмий, олово). Для этой группы металлов (кроме ртути) наиболее отчетливо проявляются неустойчивость потенциала во времени, сложный характер роста катодного осадка и другие особенности, свойственные процессу катодного выделения металлов. При промышленных плотностях тока эти металлы дают грубые осадки. Токи обмена для металлов этой группы очень велики. Так, например, ток обмена между металлической ртутью и раствором ее нитрата превышает 103 А-м-2, а между серебром и раствором нитрата серебра достигает 102 А-м~2.
Висмут, медь, цинк образуют вторую, промежуточную группу. Для нее характерно металлическое перенапряжение порядка нескольких десятков милливольт, образование более тонких осадков и меньшие, чем у металлов предыдущей группы, токи обмена. Для меди, например, ток обмена в контакте с раствором сульфата меди близок к Ю-1 А-м-2.
Наибольшим металлическим перенапряжением обладают металлы третьей группы, у которых оно достигает нескольких десятых долей вольта. Эти металлы выделяются на катоде в виде плотных тонкокристаллических осадков. Токи обмена у них малы, составляя для железа и никеля в растворах их сульфатов соответственно 10-4и 10-5А-м-2.
Данные, приведенные в табл. 22.1, относятся к обычным условиям электролиза, когда металл выделяется на поликристаллической основе и дает отложения, также имеющие поликристаллическую структуру. Поверхность таких осадков образована гранями с различными кристаллографическими индексами. В зависимости от режима электроосаждения на поверхности осадка могут преобладать те или иные грани. Поэтому важно выяснить, зависит ли металлическое перенапряжение от того, на какой грани выделяется металл. Опыты с монокристаллами ряда металлов, ориентированными по отношению к раствору различными граниями, подтвердили существование подобной зависимости (см. табл. 22.2).
Таблица 22.2. Зависимость металлического перенапряжения (В• 103) от природы грани монокристалла (при 10 Ам~2, / = 25° С)
| Индекс грани | ||||
| Металл | Раствор | |||
| (100) | (ПО) | (Ш) | ||
| РЬ | 0,5 М РЬ(СЮ4)2, | 3,0 | 3,0 | 4,4 |
| 0,5 М НС1О4 | ||||
| Sn | 0,5 М SnCl2 | 2,5 | 4,0 | _____ |
| 0,5 М НС1 | ||||
| Си | 0,5 М Си(С1О4)>, | 35 | 40 | 43 |
| 0,5 М НС1О4 | ||||
| Ni | 10 М NiCl2, | 768 | 839 | 819 |
| 0,33 М Н3ВО3 | ||||
| (рН 3,1) | ||||
Из табл. 22.1 и 22.2 следует также, что значение металлического перенапряжения в большей степени определяется природой металла, чем кристаллографической ориентацией электродной поверхности. Независимо от того, на какой из граней происходит выделение металла, перенапряжение всегда выше для никеля, чем для меди, а для меди оно всегда больше, чем для олова или свинца.
На подчиненную роль кристаллизационных факторов в явлениях перенапряжения указывают также данные по кинетике катодного выделения растворимых в ртути металлов на соответствующих амальгамах. Результаты кинетического исследования реакций обмена металлическими ионами между разбавленными амальгамами и растворами нитратов указывают на уменьшение тока обмена в следующем ряду:
TI, Pb, Cd>Cu, Zn>Ni
Характер кривых потенциал — время, полученных Гейровским методом осциллографической полярографии, показывает, что степень обратимости реакции разряда и ионизации на ртутных (точнее амальгамных) электродах уменьшается в последовательности TI, Pd, Cd, Sn, Bi, Sb, Sb, Cu
Порядок расположения металлов по степени их необратимости, а следовательно, по величине металлического перенапряжения практически не зависит от того, осаждается ли металл на твердом одноименном катоде или на разбавленной амальгаме соответствующего металла. Выделение металлов группы железа и на ртутном катоде сопровождается значительно большей поляризацией, чем у всех других металлов, приведенных в табл. 22.1. Оно протекает здесь еще менее обратимо, чем на твердых катодах. Однако эти металлы почти не способны образовывать амальгамы, и их осаждение в случае применения ртутных катодов совершается на плохо связанных между собой мелких кристаллических островках.
Роль состава раствора
Систематические исследования влияния состава раствора на кинетику электроосаждения металлов были начаты в 1917 г. Н. А. Из-гарышевым. Было установлено, что при катодном выделении металлов из растворов их простых солей существенное значение имеет природа аниона соли. Влияние природы аниона на перенапряжение и на характер образующихся осадков наблюдается для многих металлов, но наиболее сильно оно проявляется для металлов, выделение которых не сопровождается высокой поляризацией. Обычно перенапряжение уменьшается при переходе от одного аниона к другому в следующем порядке:
POJj-, №^, SO2-, C104->NH2S03~>Cl->Br->I-
Причем в том же направлении увеличивается тенденция к образованию более грубых, крупнокристаллических осадков. Влияние анионов вполне сравнимо с эффектами, связанными с кристаллографическими факторами. Так, например, замена перхлоратных растворов на сульфаминовые уменьшает перенапряжение при выделении свинца примерно в той же степени, как и переход от грани (111) к грани (ПО).
Присуствие в растворе, помимо ионов разряжающегося металла, «индифферентных» катионов увеличивает металлическое перенапряжение. Подобные эффекты наблюдались при выделении никеля, цинка, меди и других металлов. В водных растворах обычными «посторонними» катионами являются водородные ионы. Увеличение их концентрации приводит чаще всего к росту металлического перенапряжения. Значительное его повышение наблюдается в присутствии поверхностно-активных катионов типа тетразамещенного аммония.
Высокая чувствительность процесса электроосаждения металлов к чистоте растворов указывает на то, что присутствие не только электролитов, но и любых веществ, особенно обладающих поверхностно-активными свойствами, должно играть здесь существенную роль. Так, введение в ванну цинкования ничтожного количества желатины (порядка 0,005%) изменяет величину катодный поляризации и характер получающихся осадков (Н. А. Йзгарышев, П. С. Титов, 1917).
Введение в раствор небольших количеств молекулярных и ионных веществ — один из наиболее эффективных способов воздействия на ход процесса электроосаждения металлов. Многие, преимущественно органические, вещества способны увеличивать блеск осадков (блескообразователи), сглаживать их поверхность (выравниватели), и изменять другие свойства, например пористость, твердость, хрупкость, способность окклюдировать водород и т. д. (Кудрявцев, Матулис и др.).
Обнаруженная М. А. Лошкаревым адсорбционная поляризация проявляется в том, что при добавлении к раствору некоторых поверхностно-активных веществ (например, трибензиламина) изменяется скорость выделения металла на ртутном и на твердых катодах. Она становится, во-первых, меньше той, что наблюдалась до введения добавки, и, во-вторых, не зависящей в широкой области потенциалов от катодного потенциала. Однако после того как достигается определенный (обычно весьма отрицательный) потенциал, действие добавки прекращается. Скорость выделения начинает быстро расти, приближаясь к нормальному для этих условий значению, отвечающему предельному диффузионному току. Сопоставление результатов поляризационных измерений на ртутных катодах с электрокапиллярными кривыми и кривыми дифференциальной емкости (снятыми до и после введения добавки) показали, что потенциал, при котором прекращается действие добавки, совпадает с потенциалом ее десорбции (рис. 22.5). Действие добавки оказывается при этом специфическим. Одни и те же добавки или определенная их комбинация в разной степени тормозят разряд различных ионов на ртутном катоде. Явление адсорбционной поляризации используется для улучшения качества гальванических осадков при электролитическом получении сплавов.
Все эти данные относятся к тому случаю, когда металлы выделяются из растворов их простых солей. Если неорганические или органические добавки образуют комплексные соединения с выделяющимся металлом, то ход катодного процесса существенно меняется. Прежде всего образование комплексов в растворе смещает равновесный потенциал металла в отрицательную сторону за счет уменьшения концентрации его свободных ионов. Добавление вещества М. А (анионы которого способны давать комплексные соединения МАХ с ионами выделяемого металла Мг+) вызывает в растворе реакцию комплексообразования:
Мг+ +х А- = МА£"* , (22 10)
где (г—х)—заряд образующихся ионов. Реакции (22.10) отвечает константа комплексообразования
Обратная ей величина называется константой нестойкости комплекса
Константа нестойкости характеризует способность комплекса с диссоциации с регенерацией исходных ионов Mz+ и, таким образом, определяет их равновесную концентрацию.
В результате реакции комплексообразования определенная доля (юнов Мг+ (тем большая, чем ниже константа нестойкости) будет присутствовать в растворе в виде сложных ионов MA*""* и, следовательно, концентрация свободных ионов металла должна уменьшиться. Это уменьшение и, соответственно, сдвиг эбратимого потенциала электрода в отрицательную сторону будут тем значительнее, чем меньше констан-га нестойкости и чем выше концентрация добавки. Подбирая соответствующие комплексообразо-вателн и их концентрации, можно изменить равновесные потенциалы присутствующих в растворе попов различных металлов таким образом, чтобы обеспечить или их совместное осаждение в виде сплава, или наиболее полное разделение.
Появление комплексов в растворе сказывается не только на равновесных потенциалах металлов, но и на величине перенапряжения и на характере катодных осадков. При переходе от простых электролитов к комплексным обычно наблюдается повышение перенапряжения и уменьшение зернистости осадков; одновременно подавляется тенденция к образованию и росту дендритов. Так, серебро, которое при электролизе раствора его нитрата выделяется на катоде почти без поляризации и дает грубые, шероховатые осадки, из комплексных цианистых электролитов может быть получено в виде гладких тонкокристаллических отложений.
Заключение
Лаборатория электрокатализа и коррозии (зав. лабораторией — д.х.н., профессор О.А.Петрий)
В лаборатории изучается широкий круг электрокаталитических и коррозионных явлений на металлах, сплавах, оксидах и различных композиционных материалах в водных и апротонных средах. В основе подхода к этим исследованиям лежит экспериментальное и теоретическое рассмотрение процессов на микроскопическом уровне, поэтому наряду с электрохимическими методами используются и совершенствуются применительно к новым объектам и задачам методы сканирующей туннельной микроскопии и спектроскопии, эллипсометрии, а также комплекс физико-химических методов структурного анализа материалов. Разрабатываются нетрадиционные способы получения наноструктур и многофазных композиций, основанные на процессах анодной и катодной электрокристаллизации. Развиваются новые подходы к молекулярному дизайну поверхностей электродов-катализаторов.
В теоретическом плане важнейшими задачами лаборатории являются развитие теории электрокатализа, представлений о механизмах электрокаталитических процессов, а также установление закономерностей элементарного акта переноса электрона в гетерогенных системах, в первую очередь экспериментальная проверка квантово-механической теории элементарного акта с использованием нетрадиционных модельных реакций. Развиваются представления о влиянии заряда электрода на различные кинетические параметры и рассматриваются на микроскопическом уровне эффекты двойного электрического слоя в электрохимической кинетике. На основе экспериментальных исследований электродов с обновляемой поверхностью и компьютерного моделирования развиваются представления о реконструкции многокомпонентных поверхностей и о процессах поверхностной диффузии.
С целью разработки никель-металлогидридных аккумуляторов проводится широкий поиск материалов АВ5- и АВ2-типов, обратимо сорбирующих водород в электрохимических условиях.
С 1997 г. лаборатория входит вместе с Институтом физической химии РАН и кафедрами физической и коллоидной химии в состав Учебно-научного центра «Нанохимия».
Последние публикации
1. O.A.Petrii, G.A.Tsirlina, Electrochemistry of Oxide High-Temperature Superconductors, in «Adv. Electrochem. Sci. Eng.»/Ed. R.C.Alkire, H.Gerisher, D.M.Kolb and C.W.Tobias, 1997, V.5, P.61-123.
2. R.R.Nazmutdinov, G.A.Tsirlina, Yu.I.Kharkats, O.A.Petrii, M.Probst, Activation Energy of Electron Transfer between a Metal Electrode and Reagents of Nonspherical Form and Complicated Charge Distribution. Cr(EDTA) Complexes//J.Phys. Chem, 1998, V.102, P. ??-??.
3. V.A.Safonov, M.A.Choba, Yu.D.Seropegin, Peculiarities of the Electrical Double Layer Structure on Renewed Electrodes of Eutectic Alloys//Electrochim. Acta, 1997, V.42, P.1907–1914.
Литература:
Основная:
1. Дамаскин Б.Б., ПетрийО.А., Основы теоретической электрохимии,
М., Высшая школа, 1978
2. Дамаскин Б.Б. , О.А. Петрий Введение в электрохимическую кинетику, 1986 г.
3. Антропов Л.И., Теоретическая электрохимия , М.,Высшая школа, 1975 г.
4. Скорчелетти В.В., Теоретическая электрохимия ,М.. Высшая школа, 1974г.
5. Ротинян А.Л., Тихонов К.И. ,Шонина И.А. Теоретическая электрохимия, Высшая школа 1980 г.
6. Попова С.С. Анодное растворение металлов и пассивация в кислых окислительных средах.- Саратов ,Изд-во СГУ, 1984 г.
7. Кукоз Ф.И. Сборник задач по теоретической электрохимии.
8. Попова С.С. Теоретическая электрохимия. Сборник задач.
9. Левин А.И., Помосов А.В. Лабораторный практикум по теоретической электрохимии.- М: Металлургия, 1979 г.
10. Дамаскин Б.Б.Практикум по электрохимии-М: Высшая школа, 1991г.
Дополнительная
1. Феттер К., Электрохимическая кинетика, М.,Л., «Химия», 1967г.
2. Фрумкин А.Н., БагоцкийВ. С., ИофаЗ.А., Кабанов Б.Н., Кинетика электродных процессов , изд-во МГУ, 1952г.
3. Дамаскин Б.Б., Петрий О.А. , Батраков В.В., Адсорбция органических соединений на электродах. М.-Л., «Наука», 1968г.
4. Дамаскин Б.Б., Принципы современных методов изучения электорохимических реакций, Изд-во МГУ ,1965г.
5. Делахей П., Новые приборы и методы в электрохимии .,ИЛ, 1957г.
6. Делахей П., Двойной слой и кинетика электродных процессов ,»Мир», 1967г.
7. Плесков Ю.В. , Фидиновский В.Ю., Вращающийся дисковый электрод, М-Л., «Наука», 1972г.
8. Измайлов Н.А., Электрохимия растворов, М-Л.,»Химия»,1966г.
9. Корыта И., Дворжак И.,Богачкова В., Электрохимия ,»Мир», 1977г.
10. Кравцов В.И. ,Электродные процессы в растворах комплексов металлов , Изд-во ЛГУ; 1969г.
11. Мишенко К.П., Полторацкий Г.М.,Термодинамика и строение водных и неводных растворов элекролитов. М-Л., «Химия» 1976г.,1984г.
12. Латимер В., Окислительные состояния элементов, ИЛ, 1954г.
13. Методы измерения в электрохимии, т.1 и 2, Под ред.Э.Егера и А.Залкинда «Мир», 1974г.
14. Использование наглядных пособий, ТСО, вычислительной техники.
Калужский Государственный Педагогический
Университет им. К.Э. Циолковского
Дата: 2019-05-28, просмотров: 351.