Генетика человека
Учебное пособие
Владивосток
2018
УДК 575(075.8)
ББК 52.54я73 Рекомендовано к печати редакционно-издательским советом
Г34 Тихоокеанского государственного медицинского университета
Рецензенты:
Ю.В. Максимова – д.м.н., профессор, заведующий кафедрой медицинской генетики и биологии ФГБОУ ВПО «Новосибирский государственный
медицинский университет» Министерства здравоохранения
Российской Федерации
А.Б. Виноградов - д.м.н., профессор, заведующий кафедрой биологии, экологии и медицинской генетики ФГБОУ ВО «Пермский государственный медицинский университет имени академика Е.А. Вагнера» Министерства здравоохранения
Российской Федерации
Авторы:
Зенкина В.Г. – кандидат медицинских наук, доцент, заведующая кафедрой биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Солодкова О.А. - кандидат медицинских наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Божко Г.Г. – кандидат биологических наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Масленникова Л.А. - кандидат биологических наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Г34 Генетика человека: учебное пособие. – Владивосток: Медицина ДВ, 2018. – 106 с.: ил.
В учебном пособии даны основные понятия, термины, методы исследования генетики человека. Уделено внимание современным технологиям в диагностике наследственных болезней. Описаны различные наследственные болезни, симптоматика, диагностика, методы коррекции.
Учебное пособие составлено на основе Федерального государственного образовательного стандарта высшего профессионального образования по специальностям «Лечебное дело», «Педиатрия», по дисциплине «Биология».
Учебное пособие издано в помощь студентам Лечебного и Педиатрического факультетов и может быть использовано студентами всех факультетов медицинского университета.
| ОГЛАВЛЕНИЕ Введение ………………………………………………………………… Глава I. Методы изучения наследственной патологии …………………………………………………………… 1.1.Клинико-генеалогический метод …………………………………. 1.2.Близнецовый метод ………………………………......................... 1.3.Цитогенетический метод …………………………………………… 1.4.Молекулярно-цитогенетические методы ………………………… 1.5.Молекулярно-генетические методы ………………………………. 1.6.Биохимический метод ……………………………………………… 1.7.Популяционно-статистический метод …………………………… 1.8.Дерматоглифический метод ……………………………………… 1.9.Иммунологический метод ………………………………………….. 1.10.Метод генетики соматических клеток …………………………… 1.11.Методы моделирования …………………………………………... 1.12.Тестовые задания по главе I ……………………………………… Глава II. Наследственные заболевания ………………..... 2.1.Классификация наследственных болезней ………………………. 2.2.Механизм возникновения хромосомных болезней ……………..... 2.3.Клинико-цитогенетические характеристики некоторых геномных болезней ………………………………………………… 2.4.Характеристика некоторых хромосомных болезней (аббераций). 2.5.Болезни импринтинга ………………………………………………. 2.6.Наследственные болезни обмена веществ (генные заболевания).. 2.7.Тестовые задания по главе II ……………………………………..... Глава III. Общие принципы диагностики, лечения и Профилактики наследственных болезней. Медико-генетическое консультирование ………………………… 3.1.Компьтерная диагностика в генетике …………………………… 3.2.Диагностика наследственной патологии. Медико-генетическое консультирование ………………………………………………… 3.3.Пренатальная диагностика ………………………………………… 3.4.Подходы к лечению наследственных болезней ………………… 3.5.Профилактика наследственной патологии. Программа оптимального планирования семьи ……………………………… 3.6.Тестовые задания по главе III …………………………………….. Глава IV. Ситуационные задачи ……………………………. 4.1.Составление и анализ родословных……………………………… 4.2.Анализ идиограмм кариотипов человека …………………………. 4.3.Задачи на получение наследственных синдромов ……………….. 4.4.Задачи на закон Харди-Вайнберга (популяционно-статистический метод генетики человека) ……………………….. Ситуационные задачи для самостоятельного решения ……………… Эталоны ответов на тестовые задания ………………………………… Рекомендуемая литература …………………………………………….. | 5 6 7 12 14 19 21 26 28 29 30 33 34 35 40 40 41 42 45 46 48 55 60 62 65 66 71 76 78 82 82 89 91 94 97 104 105 |
ВВЕДЕНИЕ
Генетика человека – наука и фундаментальная, и прикладная. Как фундаментальная наука – это область генетики, которая изучает законы наследственности и изменчивости у самых интересных организмов – людей. Научные результаты, полученные при этом, ценны для нас не только в теоретическом отношении, но и в практическом плане. Вот почему генетика человека – это также и прикладная наука. Со времени переоткрытия законов Г. Менделя началось изучение генетических механизмов. Оно привело к расшифровке генетического кода, описанию процессов транскрипции, трансляции и функционирования белков, кодируемых определенными генами. В настоящее время уточняется тонкая структура генов, активно проводятся исследования по регуляции активности генов в ходе развития и функционирования организмов.
Генетика человека – обширная наука с неопределенными границами. Развитие различных подходов и методов привело к появлению множества отдельных специальных разделов этой науки: биохимическая генетика человека, цитогенетика, иммуногенетика, клиническая генетика, популяционная генетика, генетика поведения, генетика развития, генетика размножения, фармакогенетика и ряд других.
Близнецовый метод
Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном в 1876 году, который предложил использовать метод анализа близнецов для разграничения роли наследственности и среды в развитии различных признаков у человека. Близнецовый метод используется в генетике человека для изучения закономерностей наследования признаков в парах одно- и двуяйцевых близнецов. Позволяет выявить наследственный характер признака, определить пенетрантность аллеля, оценить эффективность действия на организм некоторых внешних факторов (лекарственных препаратов, обучения, воспитания). Исследователь работает с парами близнецов, изучая у них наличие и степень выраженности интересующего его признака, сопоставляя: монозиготных или однояйцовых близнецов с дизиготными или двуяйцовыми, партнеров в монозиготных парах между собой, данные анализа близнецовой выборки с общей популяцией. Однояйцевые близнецы развиваются из одной оплодотворенной яйцеклетки и имеют одинаковую конституцию, поэтому выявляемые между ними различия не связаны с наследственными факторами. Двуяйцевые близнецы развиваются из разных яйцеклеток, оплодотворенных разными сперматозоидами. Степень их генетического сходства такая же, как у обычных братьев и сестер. Но благодаря одновременному рождению они имеют больше общих средовых факторов. Частота рождения близнецов в популяции составляет около 1%, на долю монозиготных приходится 30%. Рождение однояйцовых близнецов часто передается по женской линии.
Результатом сравнения двух групп близнецов является расчет идентичности или конкордантности различных признаков или болезней, проявляющихся у каждого из пары близнецов К=(n/N) х100, где n-число пар близнецов, у которых признак присутствует у обоих партнеров, N-общее число обследуемых пар.
Определение степени конкордантности у близнецов имеет не только теоретическое значение, но и прямой практический выход, так как позволяет прогнозировать риск возникновения того или иного заболевания у второго партнера по близнецовой паре. С помощью близнецового метода можно оценивать проявляемость действия гена у носителей (пенетрантность), что позволяет успешно и рано включать лечебные средства для коррекции патологических нарушений.
Установление показателя дисконкордантности по уровню пенетрантности также имеет важное практическое значение, так как позволяет оценить вклад факторов среды в реализацию признака и в случае патологии принять необходимые меры профилактики в отношении второго близнеца из близнецовой пары (особенно при прогнозировании мультифакториальной патологии).
Количественной оценкой доли наследственной обусловленности признака является коэффициент наследуемости (формула Хольцингера): Н=(Км.б.-Кд.б.)/(100-Кд.б.), где Км.б., Кд.б. – конкордантность признака для моно- и дизиготных близнецов в %. Если Н>70%, решающая роль в проявлении признака принадлежит наследственным факторам. При Н<30% средовые факторы являются основными в формировании признака. При промежуточных значениях предполагается участие в контроле признака как генетических, так и средовых факторов.
Близнецовый метод в последние годы применяется редко, в основном используется специалистами по медицинской психологии.
Цитогенетический метод
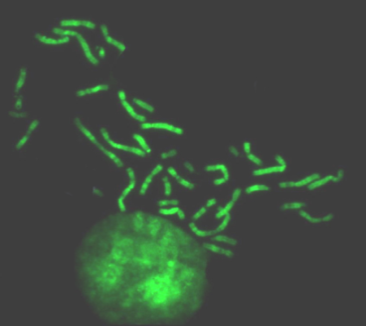
Цитогенетический метод основан на микроскопическом изучении хромосом. Хромосомы, как индивидуальные структуры становятся доступными в период деления клетки в результате значительного укорочения и утолщения. Наиболее удобной для изучения является стадия метафазы митоза, когда хромосомы наиболее спирализованы и находятся на экваторе клетки. Цитогенетический метод – основа кариотипирования, изучения идиограмм, исследования полового хроматина.
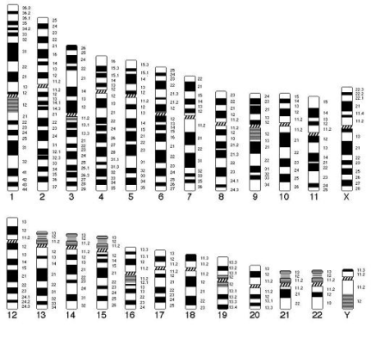
Кариотип – диплоидный набор хромосом клетки, характеризующийся количеством, величиной и формой. Систематизированное изображение кариотипа, где хромосомы пронумерованы в соответствии с их величиной, формой и расположению центромеры называют идиограммой. В 1960г. в г. Денвере, университете штата Колорадо (США) была разработана первая Международная классификация хромосом человека (Денверская классификация), согласно которой все хромосомы человека подразделили на 7 групп в зависимости от формы, размеров хромосом и расположения центромеры. Важным параметром этой классификации является центромерный индекс (ЦИ) хромосом, т.е. отношение длины короткого плеча к длине всей хромосомы (в %).
1. Группа А: 1, 2, 3 крупные метацентрические и субметацентрические хромосомы, ЦИ от 38 до 49
2. Группа В: 4, 5 крупные субметацентрические хромосомы, ЦИ 24-30
3. Группа С: 6 - 12 средние субметацентрические хромосомы, ЦИ 27-35
4. Группа Д: 13 - 15 средние акроцентрические хромосомы, ЦИ около 15
5. Группа Е: 16 - 18 мелкие субметацентрические хромосомы, ЦИ 26-40
6. Группа F: 19, 20 самые мелкие метацентрические хромосомы, ЦИ 36-46
7. Группа G: 21, 22 мелкие акроцентрические хромосомы, ЦИ 13-33
} Х – хромосома относится к группе С
} У – хромосома относится к группе G
Методы изучения хромосом:
1. Прямой метод – исследование клеток костного мозга (используется редко);
2. Непрямые методы: исследование клеток крови, фибробластов кожи, клетки абортированного плода, некропсия органов.
Наиболее удобным обьектом для медицинских генетиков являются лимфоциты периферической крови. Берут 1-2 мл крови и добавляют ее в смесь питательной среды с фитогемагглютинином (белок бобовых растений), стимулирующий деление клеток. Продолжительность культивирования составляет 48-72 часа. Затем добавляют колхицин, разрушающий нити веретена деления и прекращение митоза на стадии метафазы. Гипотонический шок вызывают гипотоническим раствором хлорида кальция или цитрата натрия, благодаря чему клетка набухает и лопается. Хромосомы фиксируют, окрашивают и микроскопируют.
Типы окраски: 1) метод Гимзе – рутинная окраска, при которой возможна только групповая идентификация хромосом и определение числовых аномалий кариотипа; 2) дифференциальное окрашивание: а) методы, выявляющие поперечную исчерченность (чередование светлых и темных поперечных полос), специфичную для каждой хромосомы – Q, G и R-окрашивание; б) методы, селективно окрашивающие определенные участки хромосом – C, T и др.
 В 1968г. Т. Касперсон предложил метод окрашивания хромосом квинакрином с последующим облучением их ультрафиолетом и индукцией флюоресценции. Оказалось, что в разных районах хромосом выявляется разное число сайтов связывания красителя, которые к тому же сильно варьировали по размерам и интенсивности свечения. Наборы флюоресцирующих полос создавали индивидуальность не только целых хромосом, но и их плеч. В результате каждую хромосому оказалось возможным идентифицировать (Q окраска).
В 1968г. Т. Касперсон предложил метод окрашивания хромосом квинакрином с последующим облучением их ультрафиолетом и индукцией флюоресценции. Оказалось, что в разных районах хромосом выявляется разное число сайтов связывания красителя, которые к тому же сильно варьировали по размерам и интенсивности свечения. Наборы флюоресцирующих полос создавали индивидуальность не только целых хромосом, но и их плеч. В результате каждую хромосому оказалось возможным идентифицировать (Q окраска).
 В 1971г. К. Шо, Э. Самнер и У. Шнедл предложили G-окраску. Препараты после предварительной щелочной обработки инкубируют в солевом растворе, а затем окрашивают красителем Романовского-Гимза. В результате появляются темные поперечные полосы. Полосам присваивают определенные номера и относительно них картируют гены. При дифференциальном окрашивании метафазных хромосом в кариотипе человека можно выявить от 200 до 400 специфических полос (бэндов). Если же вместо метафазных хромосом использовать прометафазные хромосомы, то общее число полос в кариотипе может быть увеличено до 800-1200.
В 1971г. К. Шо, Э. Самнер и У. Шнедл предложили G-окраску. Препараты после предварительной щелочной обработки инкубируют в солевом растворе, а затем окрашивают красителем Романовского-Гимза. В результате появляются темные поперечные полосы. Полосам присваивают определенные номера и относительно них картируют гены. При дифференциальном окрашивании метафазных хромосом в кариотипе человека можно выявить от 200 до 400 специфических полос (бэндов). Если же вместо метафазных хромосом использовать прометафазные хромосомы, то общее число полос в кариотипе может быть увеличено до 800-1200.
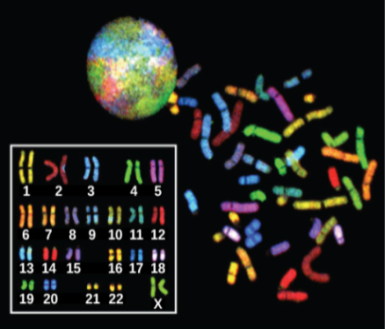 Структурные хромосомные аномалии выявляются только при дифференциальном окрашивании. Существуют также дифференцированная энзиматическая окраска, многоцветная флюоресцентная окраска (FISH – fluorescent in situ hybridization), позволяющие определить внутрихромосомные перестройки.
Структурные хромосомные аномалии выявляются только при дифференциальном окрашивании. Существуют также дифференцированная энзиматическая окраска, многоцветная флюоресцентная окраска (FISH – fluorescent in situ hybridization), позволяющие определить внутрихромосомные перестройки.
Показания к кариотипированию:
1. Множественные врожденные пороки развития
2. Привычные выкидыши
3. Недифференцированные олигофрении
4. Подозрение на семейную транслокацию
5. Пренатальная диагностика у беременной женщины после 35 лет (или мужа после 45 лет)
6. Уточнение диагноза при нарушении в системе половых хромосом
7. Нарушение репродуктивной функции неясного генеза
Природа полового хроматина
С самого раннего периода развития гистологии и цитологии в ядре были замечены интенсивно окрашивающиеся структуры. Их назвали прохромосомами, хромоцентрами, и считалось, что в этих местах хроматин проявляет положительный гетеропикноз. В настоящее время данные структуры определяются как гетерохроматин, в отличие от слабо окрашивающегося эухроматина. Впоследствии, в 1949 году, в журнале «Nature» была опубликована работа М. Бара и Ч. Бертрама, в которой ученые описали морфологические различия в нейронах самок и самцов. Открытие особых образований – глыбок гетерохроматина в интерфазных ядрах соматических клеток (телец Барра) позволило в дальнейшем использовать 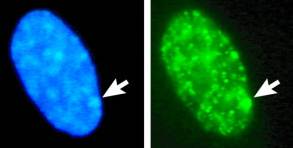 половой хроматин для решения некоторых диагностических вопросов медицинской генетики. Тельце Барра соответствует одной из двух Х-хромосом в клетках особей женского пола. В клетках у мужчин половой хроматин практически отсутствует, хотя некоторые авторы указывают на обнаружение 2-3% подобных глыбок. Первоначально для определения полового хроматина применяли биопсию кожи, но вскоре был описан метод исследования мазка слизистой оболочки ротовой полости. Суть метода в том, что штапелем делается соскоб с внутренней оболочки щеки, наносится на предметное стекло, фиксируется смесью спирта и эфира, окрашивается ацетоорсеином (крезил-виолетом или по Фельгену), с последующей микроскопией х1000.
половой хроматин для решения некоторых диагностических вопросов медицинской генетики. Тельце Барра соответствует одной из двух Х-хромосом в клетках особей женского пола. В клетках у мужчин половой хроматин практически отсутствует, хотя некоторые авторы указывают на обнаружение 2-3% подобных глыбок. Первоначально для определения полового хроматина применяли биопсию кожи, но вскоре был описан метод исследования мазка слизистой оболочки ротовой полости. Суть метода в том, что штапелем делается соскоб с внутренней оболочки щеки, наносится на предметное стекло, фиксируется смесью спирта и эфира, окрашивается ацетоорсеином (крезил-виолетом или по Фельгену), с последующей микроскопией х1000.
Существование полов, а особенно то обстоятельство, что пол животного задается различиями по половым хромосомам, ставит перед системами регуляции экспрессии генов проблему: если гены, сцепленные с Х-хромосомой, будут экспрессироваться с разными интенсивностями у особей обоих полов, то количество продуктов экспрессии в клетках самки будет в 2 раза больше, чем в клетках самца. Чтобы избежать такой ситуации существует явление компенсации доз генов. Суть явления состоит в выравнивании интенсивности экспрессии генов, расположенных на Х-хромосоме, между полами. У млекопитающих этот механизм представлен полной инактивацией одной из двух Х-хромосом в женском организме, в результате чего у самок активна только одна Х-хромосома, что эквивалентно ситуации с самцами. Мишенью для регуляции служит вся хромосома в целом, т.е. затрагиваются все промоторы на хромосоме. Хромосома переходит в состояние гетерохроматина. Возможный механизм инактивации Х-хромосомы связан с локусом Xic, где содержится ген, названный Xist. Ген Xist служит матрицей для синтеза Xist РНК (X inactive specific transcript), в которой нет открытых рамок считывания (она не является кодирующей). РНК Xist «обволакивает» данную Х-хромосому, тем самым ее инактивируя. По наличию лишнего или отсутствию тельца Барра можно диагностировать некоторые виды наследственных заболеваний, связанных с количеством половых хромосом (синдромы Клайнфельтера, Шерешевского-Тернера, полисомия по Х-хромосоме). Показаниями к исследованию полового хроматина являются: пренатальная диагностика пола, невыясненный пол новорожденного, недифференцированная олигофрения, первичная аменорея, нарушения менструального цикла и бесплодие у мужчин и женщин.
Наряду с методом определения полового хроматина, есть флюоресцирующий метод определения числа У-хромосом. Каждая У-хромосома имеет вид флюоресцирующей палочки; сколько У-хромосом в клетке, столько и флюоресцирующих палочек.
Биохимический метод
Биохимический метод используется для диагностики болезней обмена веществ, причиной которых является изменение активности определенных ферментов (генные или точковые мутации). С помощью этого метода открыто более 500 молекулярных болезней. Биохимические методы разнообразны: жидкостная хроматография, масс-спектрометрия, магнитная резонансная спектрометрия, бомбардировка быстрыми нейронами и др. Объектами биохимических методов служат любые биологические жидкости и клетки: моча, пот, плазма и сыворотка крови, форменные элементы крови, культуры клеток.
Выделяют два уровня биохимической диагностики: первичный (массовый) скрининг и уточняющий (селективный) скрининг. Массовый скрининг позволяет среди большого количества обследуемых выделить предположительно больных, имеющих какое-то наследственное отклонение от нормы (ФКУ, аномалии развития нервной трубки, врожденный гипотиреоз, адреногенитальный синдром, муковисцидоз). Селективный скрининг подразумевает уточнение диагноза у пациентов с подозрением на генные наследственные болезни.
Неонатальный скрининг - этим термином обозначают массовое обследование новорожденных для выявления ряда наследственных заболеваний. В целях своевременного выявления и их полноценной коррекции (терапии) в настоящее время в РФ осуществляется неонатальный скрининг пяти патологических состояний: фенилкетонурия (ФКУ), врожденный гипотиреоз (ГПТ), галактоземия, адреногенитальный синдром и муковисцидоз. Отметим, что ранее в РФ проводился неонатальный скрининг лишь двух заболеваний — ФКУ и врожденного ГПТ. Расширение программы неонатального скрининга с ранней диагностикой галактоземии, адреногенитального синдрома и муковисцидоза начато в 2006 году (в рамках реализации приоритетного национального проекта «Здоровье». В родильных домах России у новорожденных осуществляют забор образца крови (из пятки), которую наносят на специальную тест-полоску (тест-бланк), с последующим централизованным направлением в медико-генетическую консультацию, где проводится соответствующее исследование. При наличии подозрений на одно из перечисленных выше пяти заболеваний родители ребенка получают экстренное извещение с просьбой обратиться в медико-генетическую консультацию для повторного исследования.
Наиболее распространенными скрининг – тестами являются:
1. Проба Фелинга на ФКУ. К 2 мл мочи приливают 6 капель 10% раствора хлористого железа. Наличие сине-зелёной окраски говорит о положительном результате пробы - уровень фенилаланина больше 15 мл.
2. Проба Селиванова на фруктозурию. Несколько кристаллов резорцина растворяют в 3 мл концентрированной соляной кислоты (реактив Селиванова). К 1 объёму реактива прибавляют 2 объёма мочи, смесь подогревают на водяной бане. При наличии фруктозы смесь быстро приобретает интенсивное окрашивание, которое оценивается как положительный результат, однако, при более длительном нагревании положительную реакцию может дать и глюкоза.
3. Проба на галактозу и лактозу. К 1 мл мочи добавляют 0,5 мл концентрированного раствора аммиака и 3 капли 10% раствора едкого натрия, нагревают до кипения. Проба считается положительной при проявлении ярко-желтой окраски.
4. Проба Сулковича на кальций. К 1мл мочи добавляют 0,5 мл реактива (10,5 мл уксусной кислоты, 8,3 г щавелевой кислоты, 8,3 г сернокислого аммония на 500 мл воды). О количестве кальция судят по помутнению мочи через 1-2 минуты.
5. Проба на мукополисахариды. 1-5 капель мочи наносят на фильтрованную бумагу, высушивают. Полоска бумаги опускается в раствор толуидинового синего, с последующим отмыванием 10% раствором уксусной кислоты. Пурпурная окраска означает положительную реакцию.
Дерматоглифический метод
Индивидуальный характер папиллярных рисунков на ладонях и концевых фалангах пальцев позволяет использовать их в качестве удобных тестов для косвенной диагностики ряда наследственных заболеваний. Кожные узоры на пальцах и ладонях закладываются, начиная с третьего месяца внутриутробной жизни. К концу четвертого месяца их формирование заканчивается полностью, и в течение всей дальнейшей жизни (пре- и постнатальной) узоры остаются неизменными. Особенности узоров являются полигенными признаками, обусловленными многими генами, как наследственные факторы они подвержены мутациям в результате действия мутагенов (в первые четыре месяца жизни). Дерматоглифика подразделяется на дактилоскопию - изучение рисунка пальцев, пальмоскопию - изучение особенностей узоров ладоней и плантоскопию - изучение особенности узоров на стопах ног.
В норме частота петель, завитков и дуг (узоров подушечек пальцев) составляет соответственно 63%, 32% и 6% соответственно. Отклонения от нормы этих частот встречается у больных хромосомными болезнями. Например, при синдроме Дауна увеличивается количество петлевых узоров, при этом, частота ульнарных петель (открытый конец петли направлен в сторону мизинца) достигает 80%; при трисомии по хромосоме 18 на всех пальцах преобладают дуги, что редко обнаруживается в норме. Большое внимание уделяется сгибательным кожным складкам на ладонях. В норме у человека три крупных сгибательных складки. При синдроме Дауна в 40-60% наблюдается слияние проксимальной и средней поперечных складок с образованием одной глубокой поперечной борозды. Наличие этой складки характерно не только для синдрома Дауна, частота встречаемости этого признака в популяции 1%. В настоящее время дерматоглифические характеристики могут рассматриваться как указания на присутствие тех или иных патологий, диагностику которых надо проводить с помощью современных методов.
Иммунологический метод
Иммуногенетический метод изучает факторы иммунитета и тканевую совместимость. С помощью данного метода возможно установление причин тканевой несовместимости, определение наследования факторов иммунитета, изучение разнообразия и особенностей наследования тканевых антигенов.
Иммуногенетический метод включает иммуноэлектрофорез, серологические методики, методы ДНК-типирования и др., использующиеся для изучения групп крови, белков сыворотки крови и тканей. В настоящее время иммуногенетические методы применяются при лечении некоторых форм бесплодия, прогнозирования и ранней диагностики заболеваний с нарушением иммуногенетической совместимости.
В последнее время в качестве важного иммунологического маркёра популяционной генетики стали рассматривать главный комплекс гистосовместимости - HLA (Human Leukocyte Antigens). Антигены этой системы определяют иммунологически в лейкоцитах крови. Комплекс генов HLA компактно расположен на коротком плече хромосомы 6 (6р21.3). Локализация этой системы и протяжённость расположения её локусов на хромосоме позволили рассчитать, что комплекс составляет приблизительно 1/1000 генофонда организма. Антигены гистосовместимости участвуют в регуляции иммунного ответа организма, в поддержании иммунного гомеостаза. Благодаря своему полиморфизму и компактности локализации антигенов HLA приобрели большое значение в качестве генетического маркёра. В настоящее время обнаружено более 200 аллелей этой системы, она является наиболее полиморфной и биологически значимой из генетических систем организма человека. Нарушения различных функций главного комплекса гистосовместимости способствуют развитию ряда заболеваний, в первую очередь аутоиммунных, онкологических, инфекционных.
В соответствии с расположением комплекса HLA в хромосоме 6 различают следующие локусы: D/DR, B, C, A. Сравнительно недавно обнаружены новые локусы G, E, H, F, их биологическую роль активно изучают в настоящее время. В главном комплексе гистосовместимости выделяют три класса антигенов. Антигены I класса кодируются локусами А, В, С. Новые локусы также относятся к этому классу. Антигены II класса кодируются локусами DR, DP, DQ, DN, DO. Гены I и II классов кодируют трансплантационные антигены. Гены III класса кодируют компоненты комплемента (С2, С4а, С4b, Bf), а также синтез изоформ ряда ферментов (фосфоглюкомутазы, гликоксилазы, пепсиногена-5, 21-гидроксилазы). Наличие у человека ассоциированных с определённым заболеванием антигенов позволяет предполагать повышенную предрасположенность к данной патологии, а при некоторых корреляциях, наоборот, устойчивость к ней. Определение антигенов системы HLA проводят на лимфоцитах, выделенных из периферической крови, с помощью гистотипирующих сывороток в микролимфоцитотоксической реакции или молекулярно-генетическими методами.
Установление ассоциативных связей между болезнями и антигеном главного комплекса гистосовместимости позволяет:
· выделить группы повышенного риска развития болезни;
· определить её полиморфизм, то есть выявить группы больных с особенностями течения или патогенеза болезни; в этом же плане может проводиться анализ синтропии болезней, выяснение генетических предпосылок сочетания различных форм патологии; ассоциация с антигенами, определяющими устойчивость к заболеваниям, позволяет выявлять лиц с пониженным риском возникновения данной патологии;
· проводить дифференциальную диагностику заболеваний;
· определять прогноз;
· выработать оптимальную тактику лечения.
В связи с тем, что для большинства болезней прямой связи с антигенами главного комплекса гистосовместимости не прослеживается, для объяснения ассоциации между заболеваниями и антигенами HLA была предложена теория «двух генов», согласно которой предполагается существование гена (генов) иммунного ответа (Ir-гена), тесно связанного с антигенами HLA и генами, регулирующими иммунный ответ. Гены-протекторы определяют резистентность к заболеваниям, а гены-провокаторы - чувствительность к тем или иным болезням.
Относительный риск заболевания для лиц с соответствующим генотипом рассчитывают по формуле: x = [hp × (1 - hc)] / [hc × (1 - hp)], где hp - частота признака у больных, а hc - у лиц контрольной группы. Относительный риск показывает величину ассоциации заболевания с определённым антигеном системы HLA (даёт представление о том, во сколько раз выше риск возникновения заболевания при наличии антигена по сравнению с его отсутствием). Чем больше этот показатель у пациента, тем выше ассоциативная связь с заболеванием. Наиболее сильные ассоциативные связи выявляются для болезней с полигенным или мультифакториальным типом наследования.
Таким образом, определение антигенов главного комплекса гистосовместимости на клетках крови (лейкоцитах) позволяет выявить степень индивидуальной предрасположенности человека к определённому заболеванию, а в ряде случаев использовать результаты исследований для дифференциальной диагностики, оценки прогноза и выбора тактики лечения. Например, выявление антигенов HLA-B27 используют в дифференциальной диагностике аутоиммунных болезней. Его обнаруживают у 90-93% пациентов европеоидной расы с анкилозирующим спондилитом и синдромом Райтера. У здоровых представителей этой расы антигены HLA-B27 выявляют всего в 5-7% случаев. Антигены HLA-B27 часто обнаруживают при псориатическом артрите, хронических воспалительных заболеваниях кишечника, протекающих с сакроилеитом и спондилитом, увеите и реактивном артрите.
Методы моделирования
Как и в других разделах биологии и медицины, в генетике человека все шире применяются методы моделирования. Они разделяются на две группы: биологические и математические.
Биологическое моделирование наследственных болезней основано на законе гомологических рядов в наследственной изменчивости Н.И. Вавилова. У животных встречаются мутации, вызывающие такой же патологический эффект, как и у человека. Среди наследственных аномалий у животных встречаются гемофилия, ахондродисплазия, мышечная дистрофия, сахарный диабет и многие другие, составляющие основу наследственной патологии человека. Моделируются определенные фрагменты с целью изучения первичного механизма действия генов, патогенеза заболевания, разработки принципов их лечения.
Математическое моделирование применяется при решении задач, которые не могут быть решены путем анализа экспериментального материала. Наиболее распространено математическое моделирование в общей и популяционной генетике, для прогноза риска рождения больного ребенка (особенно для мультифакториальных заболеваний).
1.12. Тестовые задания по главе I .
Рекомендации по работе с тестовыми заданиями:
1. Необходимо внимательно прочитать задание, понять его сущность.
2. Далее, внимательно прочитать и проанализировать каждый без исключения вариант ответа.
3. Определить, какой или какие ответы правильные, отметить номера правильных ответов.
Болезни импринтинга
Некоторые гены несут, передаваемые по наследству, несут специфический «отпечаток» пола родителей. Это означает, что некоторые отцовские и материнские гены имеют различающиеся эффекты, т.е. проявляются у потомков по-разному. Это явление называется геномный импринтинг, или хромосомная память. Термин впервые предложил в 1960 г. Х. Кроуз из колумбийского университета США для описания селективной элиминации отцовских хромосом у насекомых. В начале 80-х годов XX в. началось изучение геномного импринтинга у млекопитающих после опытов на мышах, произведенных Дж. МакГратом, Д. Солтером и М. Сурани. В своих экспериментах они показали, что наследование хромосомных наборов только от одного из родителей приведет к нарушению процесса развития. Оказалось, что отцовский генетический вклад важен для развития плаценты, а материнский вклад необходим для развития тела эмбриона. Примером импринтинга целого генома у человека является истинный пузырный занос, возникающий при оплодотворении яйцеклетки, лишенной материнских хромосом, двумя сперматозоидами. В раннем эмбриогенезе таких зигот ткани собственно эмбриона не формируются, однако бурно разрастается трофобласт. В случае двойного набора материнских хромосом развивается тератома – эмбриональная опухоль.
Геномный импринтинг является формой неменделевского эпигенетического наследования, которое характеризуется дифференциальной экспрессией гена в зависимости от его родительского происхождения – матери или отца. Известно уже около 60 импринтированных генов, многие из которых оказывают существенное влияние на рост и развитие плода. Основным эпигенетическим модификатором генома является метилирование цитозиновых оснований ДНК, определяющее взаимодействие между ДНК и белками, распознающими модифицированные основания, и регулирующее экспрессию генов через механизм компактизации—декомпактизации хроматина. Нарушения моноаллельной экспрессии генов приводят к развитию особого класса наследственных заболеваний человека — болезней геномного импринтинга, которых насчитывается уже более 30.
В последнее время интенсивно изучается эффект геномного импринтинга в связи с различной патологией у человека. Наиболее убедительные данные получены при синдромах Прадера-Вилли и Ангельмана, клинически проявляющихся по-разному, в своей основе имеющих сходные молекулярно-цитогенетические изменения.
Синдром Прадера – Вилли. Делеция длинного плеча 15 хромосомы (по отцовской хромосоме). Частота встречаемости 1:10000-20000. Основные клинические проявления: мышечная гипотония, гипогонадизм, ожирение, умственная отсталость, уменьшенные размеры кистей и стоп и множественные признаки дисморфогенеза (долихоцефалия, гипертелоризм, эпикант, миндалевидный разрез глазных щелей, высокое небо). Продолжительность жизни 25-30 лет.
Синдром Ангельмана. Делеция длинного плеча 15 хромосомы (по материнской хромосоме). Частота встречаемости 1:20000. Характерные клинические проявления: приступы неконтролируемого смеха, резкие судорожные движения рук, необычная походка, хлопанье в ладоши и специфическая гримаса, частое высовывание языка, редкие зубы, задержка умственного и моторного развития, нарастает тяжесть неврологической симптоматики. Продолжительность жизни 25-30.
Пренатальная диагностика
Пренатальная диагностика – комплексная дородовая диагностика с целью обнаружения патологии на стадии внутриутробного развития. Пренатальная диагностика имеет исключительно важное значение при медико-генетическом консультировании, поскольку она позволяет перейти от вероятного к однозначному прогнозированию здоровья ребенка в семьях с генетическими осложнениями. В настоящее время пренатальная диагностика осуществляется в I и II триместрах беременности, то есть в периоды, когда в случае выявления патологии еще можно прервать беременность. На сегодня возможна диагностика практически всех хромосомных синдромов и около 100 наследственных болезней, биохимический дефект при которых установлен достоверно.
Показаниями к проведению пренатальной диагностики являются: возраст матери 35 лет; наличие в семье предыдущего ребенка с хромосомной патологией, в том числе с синдромом Дауна; перестройки родительских хромосом; наличие в семье заболеваний, которые наследуются, сцеплено с полом; синдром фрагильной Х- хромосомы; гемоглобинопатии; врожденные ошибки метаболизма; различные наследственные заболевания, диагностируемые методом сцепления с ДНК-маркерами; дефекты нервной трубки и другие.
Методы пренатальной диагностики:
1. Просеивающие – позволяют выявить женщин, имеющих повышенных риск рождения ребенка с наследственной болезнью
2. Неинвазивные – обследование плода без оперативного вмешательства
3. Инвазивные – исследование клеток эмбриона или провизорных органов
Просеивающие методы:
1. Медико-генетическое консультирование
2. Определение сывороточных маркеров: концентрация данных маркеров позволяет предположить возможные врожденные пороки развития.
- α-фетопротеин (АФП) - вырабатывается печенью плода во внутриутробном периоде. Позволяет заподозрить врожденные пороки развития нервной трубки и брюшной стенки. Концентрация АФП снижена в крови женщин, вынашивающих плод с болезнью Дауна.
- хорионический гонадотропин человека
- неконъюгированный эстриол
- ассоциированный с беременностью плазменный белок – А (PAPP)
Неинвазивные методы:
1. Скрининговое (просеивающее) ультразвуковое исследование
2. Уточняющее ультразвуковое исследование
3. Магнитно-резонансная томография (МРТ)
Основным неинвазивным методом пренатальной диагностики является ультразвуковое исследование (УЗИ), которое необходимо проводить всем беременным. Ультразвуковое сканирование плода проводят не менее двух раз во время беременности каждой женщине. Первый обзор не позднее 11-12 недели, второй - в 22-24 недели. Если есть более определенные показания для УЗИ (например, предполагаемая редукция конечностей плода), то его проведения можно начинать с 10 недели. УЗИ используется для выявления задержки роста эмбриона или плода, начиная с 6-8-ой недели беременности. Это позволяет предупредить рождение 1-3 детей (с 1000 новорожденных) с серьезными врожденными пороками развития, что составляет примерно 30% всех детей с такой патологией.
Инвазивные методы:
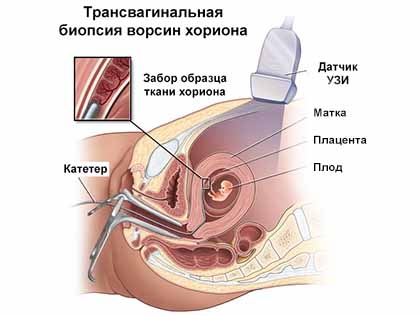 Хорионбиопсия – это взятие нескольких ворсинок из хориона плода под ультразвуковым контролем и подсчет количества хромосом в его клетках. Хорион – внезародышевый орган плода, наружная часть плодного яйца, из которого потом образуется плацента. Его клетки идентичны клеткам плода. Поэтому если количество и структура хромосом в клетках хориона нормальны, то хромосомные болезни у плода исключаются с вероятностью более 99%. Процедура проводится на 7-11 неделях беременности. Тонкой иглой через переднюю брюшную стенку или трансвагинально делается прокол, игла проходит в хорион и в нее попадает несколько ворсинок хориона. Процедура проводится в амбулаторных условиях, используются одноразовые перчатки и стерильные иглы.
Хорионбиопсия – это взятие нескольких ворсинок из хориона плода под ультразвуковым контролем и подсчет количества хромосом в его клетках. Хорион – внезародышевый орган плода, наружная часть плодного яйца, из которого потом образуется плацента. Его клетки идентичны клеткам плода. Поэтому если количество и структура хромосом в клетках хориона нормальны, то хромосомные болезни у плода исключаются с вероятностью более 99%. Процедура проводится на 7-11 неделях беременности. Тонкой иглой через переднюю брюшную стенку или трансвагинально делается прокол, игла проходит в хорион и в нее попадает несколько ворсинок хориона. Процедура проводится в амбулаторных условиях, используются одноразовые перчатки и стерильные иглы.
Плацентобиопсия – под контролем аппарата УЗИ длинной иглой делается прокол в области живота будущей мамы в сроке беременности 12-20 недель, когда плацента уже сформирована. Эта специальная игла вводится в плаценту, в результате в ней (игле) остается столбик клеток плаценты, именно их и будут исследовать.
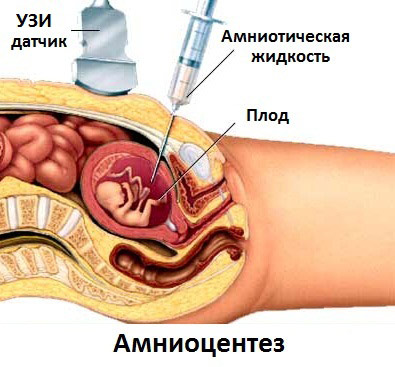 Амниоцентез - прокол плодного пузыря с целью получения околоплодной жидкости и слущенных клеток плода. Диагностическое значение метода не вызывает сомнений. Эта процедура выполняется на 15-18 неделях беременности. Риск возникновения осложнений беременности при амниоцентезе составляет 0,2%. Амниоцентез делают через брюшину под контролем УЗИ, чтобы не повредить плаценту. С амниотической полости забирают 8-10 мл жидкости. Из биохимических показателей жидкости только концентрация альфа-фетопротеина (АФП) является диагностически значимой. Основным источником диагностического материала при амниоцентезе являются клетки. Их обязательно культивируют (это длится 2-4 недели) и для цитогенетических, и для биохимических исследований. Только молекулярно-генетические варианты диагностики с помощью полимеразной цепной реакции не требуют культивирования клеток.
Амниоцентез - прокол плодного пузыря с целью получения околоплодной жидкости и слущенных клеток плода. Диагностическое значение метода не вызывает сомнений. Эта процедура выполняется на 15-18 неделях беременности. Риск возникновения осложнений беременности при амниоцентезе составляет 0,2%. Амниоцентез делают через брюшину под контролем УЗИ, чтобы не повредить плаценту. С амниотической полости забирают 8-10 мл жидкости. Из биохимических показателей жидкости только концентрация альфа-фетопротеина (АФП) является диагностически значимой. Основным источником диагностического материала при амниоцентезе являются клетки. Их обязательно культивируют (это длится 2-4 недели) и для цитогенетических, и для биохимических исследований. Только молекулярно-генетические варианты диагностики с помощью полимеразной цепной реакции не требуют культивирования клеток.
Кордоцентез - взятия крови из пуповины внутриутробного плода под контролем УЗИ. Процедуру проводят в период с 18 по 22 недели беременности. Образцы крови являются объектом для цитогенетических (культивируются лимфоциты), молекулярно-генетических и биохимических методов диагностики наследственных болезней. Кордоцентез используют для диагностики хромосомных болезней, гематологических наследственных 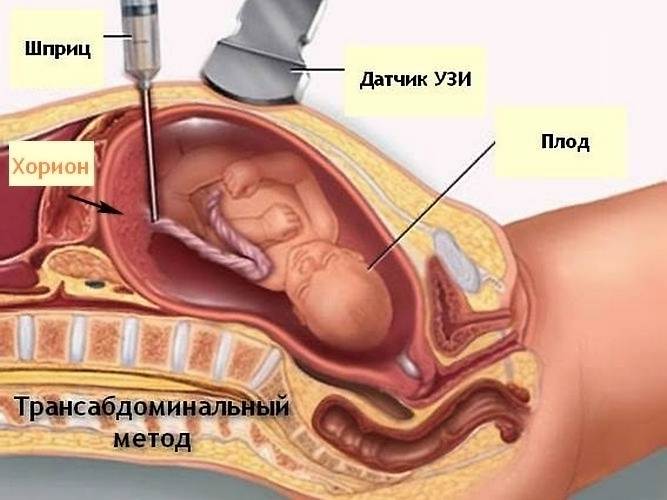 болезней (гемоглобинопатии, коагулопатии, тромбоцитопении), иммунодефицитов, гематологического статуса при резус-сенсибилизации, внутриутробных инфекций. Процедура с первой попытки успешна в 80-97% случаев. Преимущество кордоцентеза по сравнению с амниоцентезом заключается в том, что кровь является более удобным объектом для исследования, чем клетки амниотической жидкости. Лимфоциты культивируются быстрее (2-3 дня) и надежнее, чем амниоциты.
болезней (гемоглобинопатии, коагулопатии, тромбоцитопении), иммунодефицитов, гематологического статуса при резус-сенсибилизации, внутриутробных инфекций. Процедура с первой попытки успешна в 80-97% случаев. Преимущество кордоцентеза по сравнению с амниоцентезом заключается в том, что кровь является более удобным объектом для исследования, чем клетки амниотической жидкости. Лимфоциты культивируются быстрее (2-3 дня) и надежнее, чем амниоциты.
Биопсия тканей плода как диагностическая процедура осуществляется во 2-м триместре беременности под контролем УЗИ. Для диагностики тяжелых наследственных болезней кожи (ихтиоз, епидермолиз) делают биопсию кожи плода. Далее проводится патоморфологическое исследование (иногда электронно-микроскопическое). Морфологические критерии наличия наследственных болезней кожи позволяют поставить точный диагноз или уверенно отбросить его.
Фетоскопия (введение зонда и осмотр плода) при современной гибко-оптической технике не составляет большого труда. Однако метод визуального обследования плода для выявления врожденных пороков развития используется редко - только при особых показаниях. Работает на 18-23-й неделе беременности. Дело в том, что почти все врожденные пороки развития, которые можно увидеть с помощью оптического зонда, диагностируются с помощью УЗИ. Понятно, что процедура УЗИ проще и безопаснее. Для фетоскопии требуется введение зонда в амниотическую полость, что может вызвать осложнения беременности. Выкидыши отмечаются в 7-8% случаев фетоскопии. Биопсия кожи, мышц – 14-16 нед. беременности (для диагностики тяжелых наследственных болезней кожи (ихтиоза, эпидермолиза).
Задача 1.
Пробанд страдает аниридией (отсутствием радужной оболочки глаза). Он имеет две сестры, одна из которых больна аниридией. Мать пробанда здорова и вышла из благополучного по этому заболеванию рода (ее родители, бабушка и дедушка по женской линии здоровы). Отец пробанда болен. По линии отца известны больные дядя и тетя, вторая тетя здорова. У больного дяди по линии отца, женатого на здоровой женщине, больная дочь и два здоровых сына. Бабушка пробанда по линии отца, ее сестра болеют, а еще одна сестра и брат здоровые. Дедушка пробанда здоров. Прадед (отец больной бабушки) пробанда болен, а его брат здоров. Прабабушка пробанда здорова. Составить родословную. Определить тип наследования и вероятность рождения в семье пробанда больных детей, если он женится на здоровой по данному заболеванию и не имеющей больных родственников женщине.
 Решение. 1) Составляем родословную согласно правилам и символам.
Решение. 1) Составляем родословную согласно правилам и символам.
2) Анализируем, определяем тип наследования. Так как болеют мужчины и женщины в равной степени, то признак аутосомный. Больные встречаются часто, в каждом поколении, у больных детей – один из родителей имеет признак, следовательно, наследуется доминантно. Тип наследования – аутосомно-доминантный. 3) Обозначаем ген болезни - А, а ген нормы – а, и определяем зиготность пробанда и его родственников. Пробанд – гетерозиготен (Аа), так как его отец болен, а мать здорова. 4) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей.
P.: ♀ аа х ♂ Аа
G.: а А, а
F.: Аа, аа 50% вероятности рождения больных детей
Задача 2.
Пробанд - здоровая женщина, обратилась в МГК по поводу прогноза рождения у неё детей с фенилкетонурией, если она выйдет замуж за мужчину из здорового рода. Её сестра и двоюродный брат страдают фенилкетонурией, два родных брата, отец и мать (родственный брак), бабушка и дедушка со стороны матери пробанда здоровы. О семье больного двоюродного брата известно, что здоровы его родные брат, сестра, отец и мать. Отец пробанда и мать двоюродного больного брата являются родными братом и сестрой. Общий дед, бабушка отца пробанда и матери двоюродного брата, их две тетки со стороны деда здоровы. Общий прадед и прабабка со стороны бабушки матери и отца пробанда, матери больного брата здоровы. Определите тип наследования и спрогнозируйте вероятность рождения больных детей в семье пробанда.
Решение. 1) Составляем родословную согласно правилам и символам.
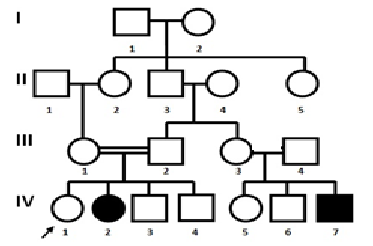 2) Анализируем, определяем тип наследования. Так как болеют мужчины и женщины в равной степени, то признак аутосомный. Больные встречаются редко, в одном поколении (горизонтальный тип наследования), у больных детей – оба родителя здоровы, следовательно, наследуется рецессивно. Тип наследования – аутосомно-рецессивный. 3) Обозначаем ген болезни - а, а ген нормы – А, и определяем зиготность пробанда и его родственников. Пробанд, может быть, как гетерозиготен (Аа), так и гомозиготен по здоровому гену (АА), так как его родители – носители патологического гена (Аа). 4) Решаем задачу и определяем вероятность рождения больных детей. Если пробанд абсолютно здоров и не является носителем гена ФКУ (АА), то выйдя замуж за такого же по генотипу мужчину, будет иметь 100% здоровых детей. Если пробанд
2) Анализируем, определяем тип наследования. Так как болеют мужчины и женщины в равной степени, то признак аутосомный. Больные встречаются редко, в одном поколении (горизонтальный тип наследования), у больных детей – оба родителя здоровы, следовательно, наследуется рецессивно. Тип наследования – аутосомно-рецессивный. 3) Обозначаем ген болезни - а, а ген нормы – А, и определяем зиготность пробанда и его родственников. Пробанд, может быть, как гетерозиготен (Аа), так и гомозиготен по здоровому гену (АА), так как его родители – носители патологического гена (Аа). 4) Решаем задачу и определяем вероятность рождения больных детей. Если пробанд абсолютно здоров и не является носителем гена ФКУ (АА), то выйдя замуж за такого же по генотипу мужчину, будет иметь 100% здоровых детей. Если пробанд
P.: ♀ Аа х ♂ Аа гетерозиготен (Аа) и ее муж здоров,
G.: А, а А, а
F.: АА, 2Аа, аа то вероятность рождения больного ребенка - 25%.
Задача 3.
Пробанд – здоровая женщина. Ее сестра также здорова, а брат страдает дальтонизмом. Мать и отец пробанда здоровы. Со стороны отца пробанда больных дальтонизмом не обнаружено. Дед и бабушка со стороны матери пробанда здоровы. Прадед со стороны бабушки болен, а прабабушка здорова. У здоровой сестры бабушки пробанда от здорового мужа родилось шесть детей: два больных сына, три дочери и сын здоровые. У одной здоровой дочери от брака со здоровым мужчиной - один больной сын, у другой дочери от брака со здоровым мужчиной – шесть детей: два больных сына и один здоровый, три здоровых дочери. Определите тип наследования, вероятность рождения у пробанда больных детей, если она выйдет замуж за своего троюродного брата - единственного сына в семье двоюродной тети.
Решение. 1) Составляем родословную согласно правилам и символам.
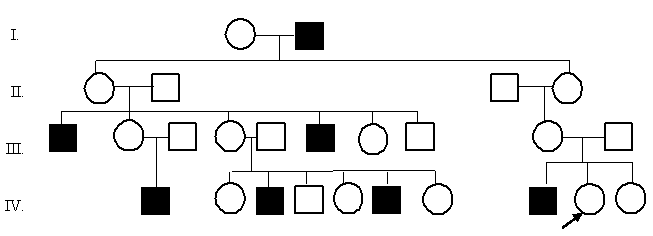 2) Анализируем, определяем тип наследования. Так как болеют только мужчины, то признак сцеплен с полом. У больных сыновей здоровые отцы, значит признак сцеплен с Х-хромосомой. Признак предается от больного отца через его фенотипически здоровых дочерей половине внуков. Больные встречаются не в каждом поколении, у здоровых родителей рождаются больные дети, следовательно, признак наследуется рецессивно. Тип наследования – Х-сцепленный рецессивный. 3) Обозначаем ген болезни - Ха, а ген нормы – ХА, и определяем зиготность пробанда и его родственников. Пробанд может быть как гетерозиготен (ХАХа), так и гомозиготен по здоровому гену (ХАХА), так как его родители здоровы, они гетерозитотны – носители патологического гена. 4) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей. Если пробанд здоров и не является носителем гена дальтонизма (ХАХА), то выйдя замуж за своего троюродного больного брата
2) Анализируем, определяем тип наследования. Так как болеют только мужчины, то признак сцеплен с полом. У больных сыновей здоровые отцы, значит признак сцеплен с Х-хромосомой. Признак предается от больного отца через его фенотипически здоровых дочерей половине внуков. Больные встречаются не в каждом поколении, у здоровых родителей рождаются больные дети, следовательно, признак наследуется рецессивно. Тип наследования – Х-сцепленный рецессивный. 3) Обозначаем ген болезни - Ха, а ген нормы – ХА, и определяем зиготность пробанда и его родственников. Пробанд может быть как гетерозиготен (ХАХа), так и гомозиготен по здоровому гену (ХАХА), так как его родители здоровы, они гетерозитотны – носители патологического гена. 4) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей. Если пробанд здоров и не является носителем гена дальтонизма (ХАХА), то выйдя замуж за своего троюродного больного брата
Р.: ♀ ХАХА х ♂ ХаУ вероятность рождения больных детей
G.: ХА Ха , У
F.: ХАУ, ХАХА составит 0%.
Если пробанд гетерозиготен (ХАХа),
P.: ♀ ХАХа х ♂ ХаУ вероятность рождения больных детей
G.: ХА, Ха Ха, У
F.: ХАХа, ХАУ, ХаХа, ХаУ составит 50%.
Задача 4.
У женщины-пробанда наблюдается синдром Гольтца (проявляется поражением кожи, дефектами скелета). У двух сестер также есть заболевание, мать пробанда больна, а отец здоров. Трое сестер матери пробанда с синдромом Гольтца, а два брата здоровы. У одной тети пробанда по линии матери от здорового мужа трое детей: дочь и сын с синдромом и здоровая дочь. Два дяди пробанда по линии матери женаты на женщинах без патологии. У одного из них два сына и дочь, у другого – две дочери и сын. Все они здоровы. Синдром Гольца имел дедушка пробанда по линии матери, а бабушка была здорова. Два брата дедушки по линии матери не имеют этого заболевания. Прабабушка (мать деда по линии матери) и прапрабабушка (мать этой бабушки) были больны, а их мужья здоровы. Составить родословную. Определите тип наследования, вероятность рождения у пробанда больных детей, если она выйдет замуж за здорового мужчину.
Решение. 1) Составляем родословную согласно правилам и символам. 2) Анализируем, определяем тип наследования. В родословной наблюдается больных женщин больше, чем мужчин, значит признак сцеплен с Х-хромосомой.
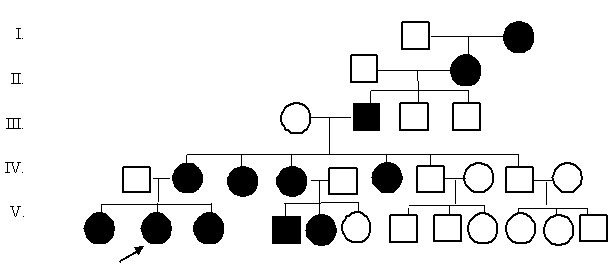 Больные встречаются в каждом поколении. У больного мужчины все дочери больны, у больных женщин страдают и девочки, и мальчики, следовательно, признак наследуется доминантно. Тип наследования – Х-сцепленный доминантный. 3) Обозначаем ген болезни – ХА, а ген нормы – Ха, и определяем зиготность пробанда и его родственников. Пробанд имеет признак и является гетерозиготным (ХАХа), так как его отец здоров (ХаУ). 4) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей, если она выйдет замуж за здорового мужчину.
Больные встречаются в каждом поколении. У больного мужчины все дочери больны, у больных женщин страдают и девочки, и мальчики, следовательно, признак наследуется доминантно. Тип наследования – Х-сцепленный доминантный. 3) Обозначаем ген болезни – ХА, а ген нормы – Ха, и определяем зиготность пробанда и его родственников. Пробанд имеет признак и является гетерозиготным (ХАХа), так как его отец здоров (ХаУ). 4) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей, если она выйдет замуж за здорового мужчину.
Р.: ♀ ХАХа х ♂ ХаУ Вероятность рождения больных детей
G.: ХА, Ха Ха, У
F.: ХАХа, ХАУ, ХаХа, ХаУ составит 50%.
Задача 5. Здоровая женщина-пробанд имеет брата, отца и деда со стороны отца с синдактилией пальцев на ногах. Мать пробанда и бабушка со стороны отца не имеют этого дефекта. Дядя со стороны отца пробанда женат на здоровой женщине. У них есть сын с синдактилией и две дочери без дефекта. Составить родословную. Определите тип наследования, вероятность рождения у пробанда больных детей, если она выйдет замуж за мужчину с синдактилией.
Решение. 1) Составляем родословную согласно правилам и символам.
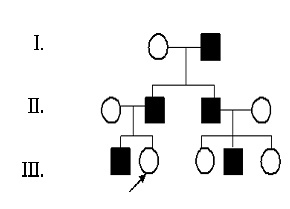 1) Болеют только мужчины, значит признак сцеплен с полом. У больных мужчин рождаются больные сыновья, следовательно, признак сцеплен с У – хромосомой. Тип наследования – голандрический. 2) Обозначаем ген болезни – У*, определяем зиготность пробанда. Пробанд не имеет этого признака, значит его генотип ХХ. 3) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей, если она выйдет замуж за больного мужчину.
1) Болеют только мужчины, значит признак сцеплен с полом. У больных мужчин рождаются больные сыновья, следовательно, признак сцеплен с У – хромосомой. Тип наследования – голандрический. 2) Обозначаем ген болезни – У*, определяем зиготность пробанда. Пробанд не имеет этого признака, значит его генотип ХХ. 3) Решаем задачу и определяем вероятность рождения в семье пробанда больных детей, если она выйдет замуж за больного мужчину.
Р.: ♀ ХХ х ♂ ХУ* Вероятность рождения больных
G.: Х Х , У*
F.: ХХ, ХУ* детей составит 50% (только мальчики).
Задача 6. Проанализируйте готовую родословную, определите тип наследования и генотип пробанда.
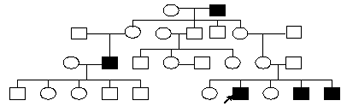
Анализируем, определяем тип наследования.
1) Так как болеют только мужчины, то признак сцеплен с полом. У больных сыновей здоровые отцы, значит признак сцеплен с Х-хромосомой. Больные встречаются не в каждом поколении, у здоровых родителей рождаются больные дети, следовательно, признак наследуется рецессивно. Тип наследования – Х-сцепленный рецессивный. 2) Обозначаем ген болезни - Ха, ген нормы – ХА, и определяем зиготность пробанда. Пробанд имеет признак, значит его генотип ХаУ.
Задача 7. Проанализируйте готовую родословную, определите тип наследования и зиготность пробанда.
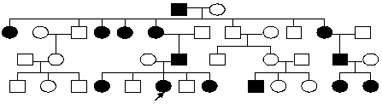
Анализируем, определяем тип наследования.
1) Болеют и мужчины, и женщины, но женщин больше, значит признак сцеплен с Х-хромосомой. Больные встречаются в каждом поколении. У больных мужчин все дочери больны, а у больных женщин больны и мальчики, и девочки в равной степени, следовательно, признак наследуется доминантно. Тип наследования – Х-сцепленный доминантный. 2) Обозначаем ген болезни - ХА, а ген нормы – Ха, и определяем зиготность пробанда. Так как мать пробанда здорова, значит она гомозиготна по нормальному аллелю (ХаХа), следовательно, пробанд, имеющий заболевание, гетерозиготен (ХАХа).
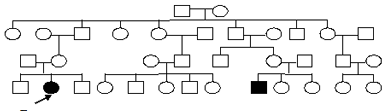 Задача 8. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Задача 8. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Анализируем, определяем тип наследования.
1) Больных в родословной мало, но болеют и мужчины, и женщины в равной степени, значит признак аутосомный. Больные встречаются не в каждом поколении. У здоровых родителей рождаются больные дети, следовательно, признак наследуется рецессивно. Тип наследования – аутосомно-рецессивный. 2) Обозначаем ген болезни - а, а ген нормы – А, и определяем зиготность пробанда. Пробанд имеет признак, значит его генотип аа.
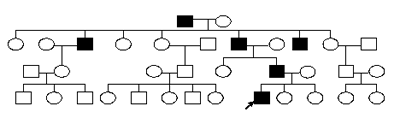 Задача 9. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Задача 9. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Анализируем, определяем тип наследования.
1) Болеют одни мужчины, значит признак сцеплен с полом. У больных мужчин рождаются только больные сыновья, следовательно, признак сцеплен с У – хромосомой. Тип наследования – голандрический. 2) Обозначаем ген болезни – У*, определяем зиготность пробанда. Пробанд имеет признак, значит его генотип ХУ*.
Задача 10. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
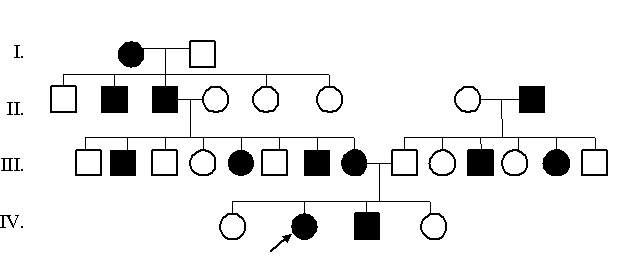
Анализируем, определяем тип наследования.
1) Болеют и мужчины, и женщины в равной степени, значит признак аутосомный. Больные встречаются в каждом поколении, наблюдается вертикальный характер передачи признака. Если один из родителей болен, то 50% детей имеет заболевание, следовательно, признак наследуется доминантно. Тип наследования – аутосомно-доминантный. 2) Обозначаем ген болезни - А, а ген нормы – а, и определяем зиготность пробанда. Пробанд имеет признак, и один из его родителей здоров, значит генотип пробанда Аа.
Задача 1.
Составить варианты возникновения синдрома дисомии по Y хромосоме (47, ХYY) с нарушением мейоза в редукционном и эквационном делении. Подробное описание механизма возникновения синдромов в главе 2.2.
Решение: Генотип 47, ХYY возможен при слиянии гамет ♀22АХ + ♂22АYY.
1) Нарушение в образовании гамет может происходить в редукционном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А+ХY 22А
å æ å æ å æ å æ
22А / 22А / 22А / 22 А / 22А!! 22А // 22А 22А
44А/!! (47, ХYY)
2) Нарушение в образовании гамет может происходить в эквационном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А+Y 22А+Х
å æ å æ å æ å æ
22А / 22А / 22А / 22А / 22А!! 22А 22А / 22А /
44А /!! (47, ХYY)
3) Такой генотип также возможен при слиянии гамет: ♀22А + ♂22АХYY
= 44АХYY. Нарушение может происходить по обеим линиям.
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А+ХY 22А
å æ å æ å æ å æ
22А / 22А / 22А // 22А 22А /!! 22А / 22А 22А
44А /!! (47, ХYY)
Задача 2.
Составить варианты возникновения синдрома Патау, трисомии по 13-15 паре – 47,ХУ+13. Получить нарушение по линии матери в редукционном делении, по линии отца в эквационном делении.
Решение: Генотип 47,ХУ+13 возможен при слиянии гамет ♀23АХ(+13) + ♂22АY.
1) Нарушение в образовании гамет может происходить в редукционном делении (по линии матери)
♀ 44АХХ ♂ 44АХY
å æ å æ
21А+Х(-13) 23А+Х (+13) 22А+У 22АХ
å æ å æ å æ å æ
21А / 21А / 23А /(+13) 23А /(+13) 22А ! 22А ! 22А / 22А /
45А /! (+13) (47, ХY+13)
Генотип 47,ХУ+13 возможен при слиянии гамет ♀22АХ + ♂23АY (+13).
2) Нарушение в образовании гамет может происходить в эквационном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А+Y 22А+Х
å æ å æ å æ å æ
22А / 22А / 22А / 22А / 23А! (+13) 21А! (-13) 22А / 22А /
45А /! (+13) (47, ХY)
Задача 3.
Составить варианты возникновения синдрома Шерешевского-Тернера, моносомии по Х-хромосоме (45,Х0), с нарушением мейоза в эквационном делении по линии матери и редукционном делении по линии отца.
Решение: Генотип 45,Х0 возможен при слиянии гамет ♀22А + ♂22АХ.
1) Нарушение в образовании гамет может происходить в эквационном делении (по линии матери)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22АХ 22А+Х 22А+У
å æ å æ å æ å æ
22А / 22А / 22А // 22 А 22А / 22А / 22А! 22А !
44А / (45, Х0)
Генотип 45,Х0 возможен при слиянии гамет ♀22АХ + ♂22А.
2) Нарушение в образовании гамет может происходить в редукционном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А 22А+ХУ
å æ å æ å æ å æ
22А / 22А / 22А / 22А / 22А 22А 22А /! 22А /!
44А / (45, Х0)
Задача 4.
Составить варианты возникновения синдрома Эдвардса, трисомии по 16-18 паре – 47,ХY+16. Получить нарушение по линии матери в эквационном делении и по линии отца в редукционном делении.
Решение: Генотип 47,ХХ+16 возможен при слиянии гамет ♀23АХ(+16) + ♂22АY.
1) Нарушение в образовании гамет может происходить в эквационном делении (по линии матери)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 22А+Х 22АУ
å æ å æ å æ å æ
22А / 22А / 21А / (-16) 23А /(+16) 22А / 22А / 22А! 22А!
45А /! (+16) (47, ХY+16)
Генотип 47,ХХ+16 возможен при слиянии гамет ♀22АХ + ♂23АY (+16).
2) Нарушение в образовании гамет может происходить в редукционном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А+Х 22А+Х 23А+Y(+16) 21А+Х (-16)
å æ å æ å æ å æ
22А / 22А / 22А / 22А / 23А! (+13) 23А! 21А / 21А /
45А /! (+16) (47, ХY+16)
Задача 5.
Составить несколько разных вариантов возникновения синдрома тетраплоидии – 92,ХХХХ.
Решение: Генотип 92,ХХХХ возможен при слиянии гамет ♀44АХХ + ♂44АХХ.
1) Нарушение в образовании гамет может происходить одновременно в редукционном делении (по линии матери) и в эквационном делении по линии отца
♀ 44АХХ ♂ 44АХY
å æ å æ
0А 44А+ХХ 22А+Х 22АУ
å æ å æ å æ å æ
0А 0А 44А // 44 А // 44А // 0А 22А! 22А!
88А //// (92, ХХХХ)
2) Нарушение в образовании гамет может происходить в эквационном делении (по линии матери) и в эквационном делении (по линии отца)
♀ 44АХХ ♂ 44АХY
å æ å æ
22А +Х 22А+Х 22А+Х 22АУ
å æ å æ å æ å æ
22А / 22А / 0А 44А // 44А // 0А 22А! 22А!
88А //// (92, ХХХХ)
4.4. Задачи на закон Харди-Вайнберга (популяционно-статистический метод генетики человека)
Задача 1.
Алкаптонурия (аутосомно-рецессивный признак) характеризуется окрашиванием суставных хрящей в оранжевый цвет и быстрым потемнением подщелоченной мочи. В возрасте после 40 лет при этой аномалии развивается упорный полиартрит, приводящий к тугоподвижности суставов. Заболевание встречается с частотой 1:100000. Вычислите частоту встречаемости гетерозигот в популяции.
Решение.
1) p2+2pq+q2=1, где p2 – частота встречаемости доминантных гомозигот (АА), q2 – частота встречаемости рецессивных гомозигот (аа), 2pq – частота встречаемости гетерозигот (Аа). Так как алкаптонурия – аутосомно-доминантное заболевание, то больные люди в популяции составляют q2, следовательно, q2=1/100000=0,000001, q=√q2=0,001
2) р+q=1, р=1-q, р=1-0,001=0,999 – частота доминантного гена
3) 2pq=2х0,999х0,001=0,0019 – частота встречаемости гетерозигот в популяции.
Задача 2.
Аниридия (отсутствие радужной оболочки) является аутосомно-доминантным признаком. Среди европейцев эта аномалия встречается с частотой 1:10000. Определите частоту доминантного и рецессивного генов по этому признаку.
Решение.
1) Аниридия – аутосомно-доминантное заболевание, значит больные люди в популяции составляют p2+2pq (АА+Аа), следовательно, р2+2pq=1/10000=0,0001
2) q2=1-(p2+2pq), q2=1-0,0001=0,9999 – частота встречаемости рецессивных гомозигот (здоровые люди). q=√q2=0,99994 – частота рецессивного гена в популяции
3) р+q=1, р=1-q, р=1-0,99994=0,00006 – частота доминантного гена в популяции.
Задача 3.
В одной изолированной популяции человека насчитывается примерно 16% людей, имеющих резус-отрицательную кровь (рецессивный признак). Установить частоту встречаемости гетерозиготных носителей гена резус-отрицательной крови.
Решение.
1) q2=0,16 – частота встречаемости рецессивных гомозигот (резус-отрицательные люди). q=√q2=0,4 – частота рецессивного гена в популяции
2) р+q=1, р=1-q, р=1-0,4=0,6 – частота доминантного гена
3) 2pq=2х0,4х0,6=0,48 – частота встречаемости гетерозигот.
Задача 4.
Глухонемота связана с врожденной глухотой, которая препятствует нормальному усвоению речи. Тип наследования заболевания аутосомно-рецессивный. Средняя частота заболевания колеблется по разным странам. Для европейских стран она равна приблизительно 2:10000. Определите возможное число гетерозиготных по глухоте людей в районе, включающем 8000000 жителей.
Решение.
1) q2=2/10000=0,0002 – частота встречаемости рецессивных (аа) гомозигот (больные люди). q=√q2=0,0141 – частота рецессивного гена в популяции
2) р+q=1, р=1-q, р=1-0,0141=0,9859 – частота доминантного гена в популяции
3) 2pq=2х0,9859х0,0141=0,0278 – частота встречаемости гетерозигот
4) Число гетерозиготных по глухоте людей в районе, включающем 8000000 жителей Аа=0,0278х8000000=222400человек.
Задача 5.
Муковисцидоз наследуется по аутосомно-рецессивному типу. Географические и этнические различия в частоте муковисцидоза и вариантах мутаций гена очень значительны. Муковисцидоз редко встречается в восточных популяциях и у африканского чернокожего населения (1:100 000). В Европе частота заболевания составляет в среднем 1:2500 новорожденных. Определите процент гетерозиготных носителей гена муковисцидоза в Европе.
Решение.
1) q2=1/2500=0,0004 – частота встречаемости рецессивных (аа) гомозигот (больные люди). q=√q2=0,02 – частота рецессивного гена в популяции
2) р+q=1, р=1-q, р=1-0,02=0,98 – частота доминантного гена в популяции
3) 2pq=2х0,02х0,98=0,0784 (7,84%) – частота встречаемости гетерозигот.
Ситуационные задачи для
самостоятельного решения
Задача 1.
Молодожены нормально владеют правой рукой. В семье женщины было ещё две сестры, нормально владеющие правой рукой, и три брата - левши. Мать женщины – правша, отец – левша. У отца есть сестра и брат левши и сестра и брат правши. Дед по линии отца правша, бабушка – левша. У матери женщины есть два брата и сестра – все правши. Мать мужа – правша, отец – левша. Бабушки и дедушки со стороны матери и отца нормально владели правой рукой. Определите тип наследования и вероятность рождения в этой семье леворуких детей.
Задача 2.
Пробанд страдает ночной слепотой. Два его брата также больны. По линии отца пробанда, страдающих ночной слепотой не было. Мать пробанда больна. Две сестры и два брата матери пробанда здоровы. Они имеют только здоровых детей. По материнской линии дальше известно: бабушка больна, дед здоров; прадедушка (отец бабушки) страдал ночной слепотой, сестра и брат прадедушки были больны; прапрадедушка (отец прадедушки) болен, его брат, имеющий больную дочь и двух больных сыновей, также болен. Жена пробанда, ее родители и родственники здоровы. Определите тип наследования и вероятность рождения больных детей в семье пробанда.
Задача 3.
У пробанда в роду наблюдается дефект эмали зубов. Родители пробанда дефекта не имели, однако со стороны родственников отца это часто наблюдалось. Дед и его брат со стороны отца пробанда имели дефект эмали. Сын брата деда пробанда не имел дефекта, а дочь, внучка с ее стороны имели темные зубы, внук – со здоровыми зубами. У деда со стороны отца пробанда пятеро детей, из которых три дочери, в том числе две – однояйцевые близнецы имеют дефект, а два сына нет. У одной из однояйцевых близнецов - больной сын и здоровая дочь. Какова вероятность появления дефекта у детей пробанда, если она выйдет замуж за своего двоюродного брата, имеющего этот дефект?
Задача 4.
Ш. Ауэрбах (1969) приводит следующую родословную по шестипалости. Две шестипалые сестры Маргарет и Мэри вышли замуж за нормальных мужчин. В семье Маргарет было пятеро детей: Джеймс, Сусанна, Дэвид - шестипалые, Элла и Ричард - пятипалыми. В семье Мэри была единственная дочь Джейн с нормальным строением руки. От первого брака Джеймса с нормальной женщиной родилась шестипалая дочь Сара, от второго брака тоже с нормальной женщиной у него было шесть детей: одна дочь и два сына - пятипалые, две дочери и один сын - шестипалые. Элла вышла замуж за нормального мужчину. У них было два сына и четыре дочери - все пятипалые. Дэвид женился на нормальной женщине. Единственный их сын Чарльз оказался шестипалым. Ричард женился на своей двоюродной сестре Джейн. Две их дочери и три сына были пятипалыми. Определите тип наследования и вероятность рождения шестипалых детей в случаях: 1) брака нормальной дочери Джеймса с одним из сыновей Ричарда; 2) брака Сары с сыном Дэвида.
Задача 5.
Пробанд имеет белый локон в волосах надо лбом. Брат пробанда без локона. По линии отца пробанда аномалии не отмечено. Мать пробанда с белым локоном. Она имеет трех сестер, две из которых с белым локоном, одна без локона. У одной из трех теток со стороны матери сын с локоном и дочь без локона, у второй - сын и дочь с локоном, и дочь без локона. Третья тетка пробанда со стороны матери (без локона) имеет двух сыновей и одну дочь без локона. Дед пробанда по линии матери и двое его братьев имели белые локоны, а ещё двое были без локонов. Прадед и прапрадед также имели локон надо лбом. Определите тип наследования, зиготность членов родословной и вероятность рождения детей с белым локоном надо лбом в случае, если пробанд вступит в брак со своей двоюродной сестрой, имеющей этот локон.
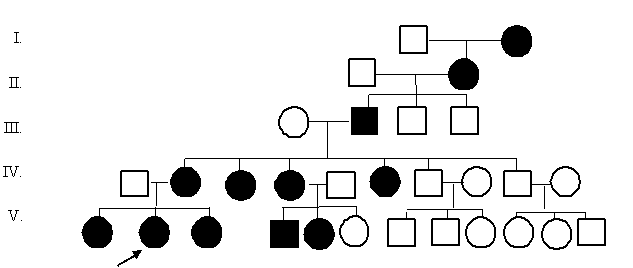 Задача 6. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Задача 6. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
Задача 7. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
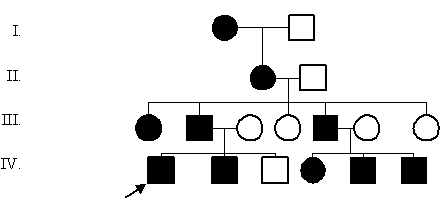
Задача 8. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
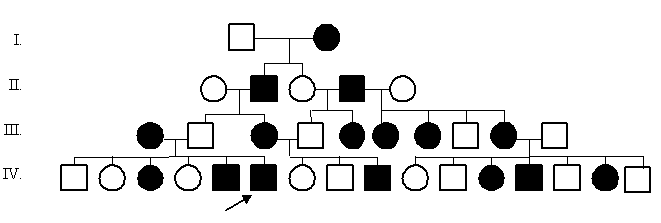
Задача 9. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
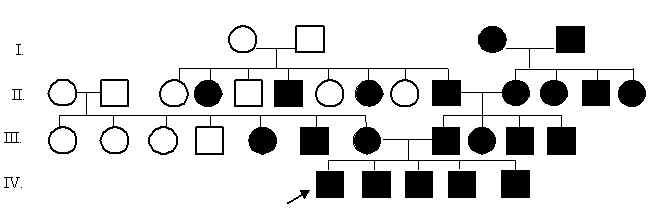
Задача 10. Проанализируйте готовую родословную, определите тип наследования и зиготность членов родословной.
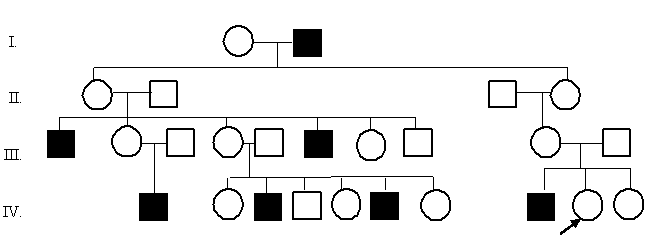
Задача 1.
Составить варианты возникновения синдрома трисомии по 8 паре хромосом – 47,ХY+8. Получить нарушение по линии матери в эквационном делении и по линии отца в редукционном делении.
Задача 2.
Составить несколько разных вариантов возникновения синдрома трисомии по 14 паре хромосом – 47, ХХ+14.
Задача 3.
Составить все возможные варианты возникновения синдрома Клайнфельтера – 47,ХХУ.
Задача 4.
Составить все возможные варианты возникновения синдрома Клайнфельтера – 49,ХХХУУ.
Задача 5.
Составить все возможные варианты возникновения синдрома тетрасомии по Х-хромосоме – 48,ХХХХ.
Задачи на закон Харди-Вайнберга (популяционно-статистический метод генетики человека)
Задача 1.
В одном из американских городов, в части, представляющей изолят из итальянских переселенцев, в период с 1928 по 1942 гг. среди 26000 новорожденных 11 оказались с тяжелой формой талассемии - генотип ТТ (К. Штерн, 1965). Определите число гетерозигот среди итальянских переселенцев данного города.
Задача 2.
Соответствуют ли формуле Харди-Вайнберга следующие соотношения: а) 239АА : 79Аа : 6аа; б) 400АА : 110Аа : 4аа?
Задача 3.
Подагра встречается у 2% людей и обусловлена аутосомно-доминантным геном. У женщин ген подагры чаще не проявляется, а у мужчин пенетрантность его равна 20% (З.П. Эфроимсон, 1968). Определите генетическую структуру популяции по анализируемому признаку, исходя из этих данных.
Задача 4.
В городе установившимся составом населения в течение 4 лет среди 25000 новорожденных зарегистрировано 2 больных фенилкетонурией, которая наследуется по аутосомно-рецессивному гену. Определите количество гетерозигот по фенилкетонурии среди населения данного города, составляющим 6000000 человек.
Задача 5.
Галактоземия наследуется как аутосомно-рецессивный признак. Частота возникновения разных типов галактоземии вариабельна. Общая частота заболевания в Европе может составлять 1:180 000. Определить частоту встречаемости гетерозигот в популяции.
ЭТАЛОНЫ ОТВЕТОВ НА ТЕСТОВЫЕ ЗАДАНИЯ
Глава 1: 1-2; 2-2; 3-1; 4-3; 5-2; 6-1,2,5; 7-2,4; 8-2; 9-3; 10-1; 11-3; 12-1,2,3,4,5; 13-1,2,3; 14-3; 15-1,2; 16-1,2,3,4,5; 17-4; 18-3; 19-1,2,3,4; 20-1; 21-2,3; 22-4; 23-2
Глава 2: 1-1,3; 2-1,2,4,5; 3-1,2; 4-3; 5-2,3; 6-1; 7-1,4; 8-4; 9-3; 10-1; 11-1; 12-3,4; 13-1,2; 14-1; 15-3; 16-2,3; 17-3; 18-1; 19-1,2,3; 20-2; 21-3; 22-3; 23-2,4; 24-4;25-1,2
Глава 3: 1-1; 2-2; 3-2; 4-3; 5-1; 6-2; 7-2; 8-1,3,4; 9-3; 10-3; 11-1,2,3; 12-1; 13-2,3; 14-2; 15-1,2; 16-4;17-1,2,3;18-1,2,3,4
Рекомендуемая литература
а) Основная
1. Чебышев Н.В. Биология: учебник - М.: Академия, 2014, 416 с.
2. Ярыгин В.Н. Биология: учебник (в 2 т.) Том 1 - М.: ГЭОТАР-Медиа, 2015, 736 с.
б) Дополнительная
1. Генетика человека с основами медицинской генетики: учебник / Е.К. Хандогина, И.Д. Терехова, С.С. Жилина и др. - М.: ГЭОТАР-Медиа, 2017, 192 с.
2. Жимулев И.Ф. Общая и молекулярная генетика: учеб. пособие – Новосибирск: Сиб. унив. изд-во, 2007, 479 с.
3. Иванов В.И. Генетика: учебник для вузов – М.: ИКЦ «Академкнига», 2006, 638с.
4. Мутовин Г.Р. Клиническая генетика. Геномика и протеомика наследственной патологии: учебное пособие - М.: ГЭОТАР-Медиа, 2010, 832 с.
5. Наследственные болезни: национальное руководство / Под ред. Н.П. Бочкова, Е.К. Гинтера, В.П. Пузырева - М.: ГЭОТАР-Медиа, 2012, 936 с. (Серия национальные руководства)
6. Новиков П.В. Семиотика наследственных болезней у детей (симптом-синдром-болезнь) – М.: «Триада-Х», 2009, 432с.
Рисунки заимствованы из открытых источников.
Учебное пособие
Зенкина Виктория Геннадьевна
Солодкова Оксана Алексеевна
Божко Галина Георгиевна
Масленникова Любовь Андреевна
Генетика человека
Учебное пособие
Владивосток
2018
УДК 575(075.8)
ББК 52.54я73 Рекомендовано к печати редакционно-издательским советом
Г34 Тихоокеанского государственного медицинского университета
Рецензенты:
Ю.В. Максимова – д.м.н., профессор, заведующий кафедрой медицинской генетики и биологии ФГБОУ ВПО «Новосибирский государственный
медицинский университет» Министерства здравоохранения
Российской Федерации
А.Б. Виноградов - д.м.н., профессор, заведующий кафедрой биологии, экологии и медицинской генетики ФГБОУ ВО «Пермский государственный медицинский университет имени академика Е.А. Вагнера» Министерства здравоохранения
Российской Федерации
Авторы:
Зенкина В.Г. – кандидат медицинских наук, доцент, заведующая кафедрой биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Солодкова О.А. - кандидат медицинских наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Божко Г.Г. – кандидат биологических наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Масленникова Л.А. - кандидат биологических наук, доцент кафедры биологии, ботаники и экологии (Тихоокеанский государственный медицинский университет, Владивосток)
Г34 Генетика человека: учебное пособие. – Владивосток: Медицина ДВ, 2018. – 106 с.: ил.
В учебном пособии даны основные понятия, термины, методы исследования генетики человека. Уделено внимание современным технологиям в диагностике наследственных болезней. Описаны различные наследственные болезни, симптоматика, диагностика, методы коррекции.
Учебное пособие составлено на основе Федерального государственного образовательного стандарта высшего профессионального образования по специальностям «Лечебное дело», «Педиатрия», по дисциплине «Биология».
Учебное пособие издано в помощь студентам Лечебного и Педиатрического факультетов и может быть использовано студентами всех факультетов медицинского университета.
| ОГЛАВЛЕНИЕ Введение ………………………………………………………………… Глава I. Методы изучения наследственной патологии …………………………………………………………… 1.1.Клинико-генеалогический метод …………………………………. 1.2.Близнецовый метод ………………………………......................... 1.3.Цитогенетический метод …………………………………………… 1.4.Молекулярно-цитогенетические методы ………………………… 1.5.Молекулярно-генетические методы ………………………………. 1.6.Биохимический метод ……………………………………………… 1.7.Популяционно-статистический метод …………………………… 1.8.Дерматоглифический метод ……………………………………… 1.9.Иммунологический метод ………………………………………….. 1.10.Метод генетики соматических клеток …………………………… 1.11.Методы моделирования …………………………………………... 1.12.Тестовые задания по главе I ……………………………………… Глава II. Наследственные заболевания ………………..... 2.1.Классификация наследственных болезней ………………………. 2.2.Механизм возникновения хромосомных болезней ……………..... 2.3.Клинико-цитогенетические характеристики некоторых геномных болезней ………………………………………………… 2.4.Характеристика некоторых хромосомных болезней (аббераций). 2.5.Болезни импринтинга ………………………………………………. 2.6.Наследственные болезни обмена веществ (генные заболевания).. 2.7.Тестовые задания по главе II ……………………………………..... Глава III. Общие принципы диагностики, лечения и Профилактики наследственных болезней. Медико-генетическое консультирование ………………………… 3.1.Компьтерная диагностика в генетике …………………………… 3.2.Диагностика наследственной патологии. Медико-генетическое консультирование ………………………………………………… 3.3.Пренатальная диагностика ………………………………………… 3.4.Подходы к лечению наследственных болезней ………………… 3.5.Профилактика наследственной патологии. Программа оптимального планирования семьи ……………………………… 3.6.Тестовые задания по главе III …………………………………….. Глава IV. Ситуационные задачи ……………………………. 4.1.Составление и анализ родословных……………………………… 4.2.Анализ идиограмм кариотипов человека …………………………. 4.3.Задачи на получение наследственных синдромов ……………….. 4.4.Задачи на закон Харди-Вайнберга (популяционно-статистический метод генетики человека) ……………………….. Ситуационные задачи для самостоятельного решения ……………… Эталоны ответов на тестовые задания ………………………………… Рекомендуемая литература …………………………………………….. | 5 6 7 12 14 19 21 26 28 29 30 33 34 35 40 40 41 42 45 46 48 55 60 62 65 66 71 76 78 82 82 89 91 94 97 104 105 |
ВВЕДЕНИЕ
Генетика человека – наука и фундаментальная, и прикладная. Как фундаментальная наука – это область генетики, которая изучает законы наследственности и изменчивости у самых интересных организмов – людей. Научные результаты, полученные при этом, ценны для нас не только в теоретическом отношении, но и в практическом плане. Вот почему генетика человека – это также и прикладная наука. Со времени переоткрытия законов Г. Менделя началось изучение генетических механизмов. Оно привело к расшифровке генетического кода, описанию процессов транскрипции, трансляции и функционирования белков, кодируемых определенными генами. В настоящее время уточняется тонкая структура генов, активно проводятся исследования по регуляции активности генов в ходе развития и функционирования организмов.
Генетика человека – обширная наука с неопределенными границами. Развитие различных подходов и методов привело к появлению множества отдельных специальных разделов этой науки: биохимическая генетика человека, цитогенетика, иммуногенетика, клиническая генетика, популяционная генетика, генетика поведения, генетика развития, генетика размножения, фармакогенетика и ряд других.
Дата: 2018-11-18, просмотров: 566.