Капельный анализ неорганических веществ
Цель и задачи работы
Овладеть приемами выполнения качественных аналитических реакций капельным способом. Освоить методику дробного качественного анализа, обнаружения катионов и анионов в многокомпонентных средах с помощью специфических реакций. Произвести анализ объекта неизвестного состава.
Программа работы
1.2.1. Предварительная подготовка пробы
Анализ твердых объектов
Пробу измельчают в мельнице или растиранием в ступке. Берут навеску 0,1-0,5 г и растворяют в соответствующем, в зависимости от предполагаемого химического состава, растворителе (вода, растворы кислот и щелочей).
1.2.1.2. Анализ металлов и сплавов (безстружковый метод)
На очищенную поверхность образца помещают 2-3 капли азотной кислоты (1:1). После прекращения реакции, образовавшийся на поверхности металла раствор разбавляют дистиллированной водой (2-3 капли) и переносят с помощью капилляра или пипетки в микропробирку. Операцию повторяют 2-3 раза. Собранный раствор подвергают непосредственно анализу.
Анализ жидких сред
В зависимости от компонентного состава пробы, при необходимости, по специальным методикам проводят операции маскирования, осаждения и отделения органических соединений и отдельных ионов, мешающих анализу, или выделения открываемых ионов.
Выполнение реакций обнаружения анионов
Обнаружение карбонатов.
|
| |
| Рис.1.1. Устройство для определения CO2 и летучих кислот. 1 пробка; 2 пробирка; 3 стеклянная палочка с шариком; 4 анализируемый раствор. |
Одну-две капли анализируемого раствора или кусочек твёрдого вещества обрабатывают в приборе тремя каплями 1 М серной кислоты. Шарик пробки опускают в реагент и устанавливают на место. Капля реагента на шарике обесцвечивается немедленно или через короткое время в зависимости от количества выделяющегося оксида углерода (IV). Чтобы избежать неправильных выводов вследствие влияния углекислого газа воздуха, необходимо параллельно проводить холостой опыт в таком же приборе.
Предел обнаружения: 4 мкг.
Реагент: смешивают 1 см3 0,05 М раствора карбоната натрия с 2 см3 0,5% (масс) этанольного раствора фенолфталеина и доводят до 10 см3 дистиллированной водой.
Обнаружению мешают летучие кислоты, влияние которых устраняется по специальным методикам.
Обнаружение нитратов
На предметное стекло вместе с каплей анализируемого раствора помещают кристаллик сульфата железа (II). Добавляют каплю концентрированной серной кислоты так, чтобы она стекала сбоку. В присутствии нитратов вокруг кристаллика образуется коричневое кольцо.
Предел обнаружения: 2,5 мкг.
Обнаружению мешают нитриты, иодиды, бромиды, циан иды, цианоферраты, хроматы, сульфиты, тиосульфаты, иодаты и др.
Обнаружение ортофосфатов
На фильтровальную бумагу наносят каплю анализируемого раствора и высушивают. Затем бумагу погружают или опрыскивают реагентом I и через 30 с реагентом II. Фосфаты образуют синее пятно.
Предел обнаружения: 0,1 мкг.
Реагенты: метилвиолет, водный раствор, w = 1% (масс); в 150 см3 растворяют 12 г молибдата аммония и подкисляют 35 см3 10 М раствором хлороводородной кислоты.
Обнаружение силикатов
На фильтровальную бумагу помещают каплю анализируемого раствора и каплю первого реагента. Затем к раствору добавляют каплю второго реагента, после чего бумагу некоторое время держат над аммиаком. Появление голубого окрашивания свидетельствует о присутствии в растворе кремниевой кислоты.
Предел обнаружения: 1 мкг.
Реагенты: в 150 см растворяют 12 г молибдата аммония и подкисляют 35 см3 10 М раствором хлороводородной кислоты; бензидин, водный раствор, w = 0,1% (масс).
Обнаружению мешают фосфаты.
Обнаружение сульфидов
К 1-2 каплям анализируемого раствора, помещенным на предметное стекло добавляют 2-3 капли реагента. В присутствии сульфидов выпадает черный осадок сульфида свинца.
Предел обнаружения: 1,8 мкг.
Регент: ацетат свинца, водный раствор, С=1 моль/дм3.
Обнаружению мешают ионы, образующие со свинцом нерастворимые соли.
Обнаружение сульфатов
На предметном стекле смешивают три капли анализируемого раствора с каплей насыщенного раствора перманганата калия. Каплю полученной смеси наносят на фильтровальную бумагу, пропитанную хлоридом бария, и выдерживают ее 7-8 мин над печью при 70-80o С. Избыток хлорида бария удаляют промыванием в воде в течение 1 мин. Бумагу помещают в 1 М раствор щавелевой кислоты для разрушения 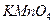 и
и 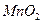 . Если в анализируемом растворе присутствует сульфат — ион, то на бумаге остается яркое фиолетовое пятно.
. Если в анализируемом растворе присутствует сульфат — ион, то на бумаге остается яркое фиолетовое пятно.
Предел обнаружения: 2,5 мкг.
Реагент: бумага, пропитанная 0,5 М раствором хлорида бария и высушенная.
Обнаружению мешают сульфид-, сульфит-, роданид-, селенат- и селенит-ионы.
Обнаружение хлоридов
Используется прибор (рис.1.1). Несколько крупинок образца помещают в пробирку прибора или же каплю испытуемого раствора выпаривают в ней. Затем добавляют две капли концентрированной азотной кислоты. На стеклянный шарик пробки наносят реагент и закрывают пробирку. Прибор нагревают на асбестовой сетке до появления первых пузырьков. При малых количествах хлорид-ионов, чтобы увидеть помутнение реагента, каплю переносят на черную фарфоровую пластинку.
Предел обнаружения: 1 мкг хлороводородной кислоты.
Реагент: нитрат серебра, водный раствор, С = 0,1 моль/дм3.
Обнаружению мешают иодиды и бромиды.
Результаты работ 1.2.2, 1.2.3 и анализ объекта неизвестного состава вносят в таблицу 1.1:
Результаты обнаружения неорганических ионов.
Таблица 1.1
| № п/п | Обнаруживаемый ион | Реагент | Внешний эффект | Вывод | |
| Стандартный раствор | Анализ | ||||
1.3. Вопросы для самоподготовки
1. В чем принципиальное отличие дробного метода анализа от систематического? Каковы преимущества и недостатки методов друг перед другом?
2. Какой метод, дробный или систематический, следует применить при обнаружении нескольких ионов в смеси, состав которой приблизительно известен?
3. Назовите критерии оценки аналитических реакций.
4. Какие реакции называются общими, специфическими, частными?
5. Дайте определения понятиям "качественная реакция", "селективность реакции", "предел обнаружения".
6. Назовите признаки по которым классифицируются аналитические реакции.
7. Какова аналитическая классификация катионов по сероводородному методу анализа?
8. На чем основано деление катионов и анионов на аналитические группы в систематическом анализе?
9. Что такое групповой реагент? Каким требованиям он должен отвечать? Как проверить полноту осаждения ионов?
10. Каков порядок осаждения катионов в систематическом ходе анализа? Почему анализ начинают с обнаружения иона 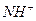 ?
?
11. Назовите групповые реагенты аналитических групп по сероводородному методу.
12. Что такое маскирование ионов? В каких случаях эта операция применяется?
13. Какие методы и способы группового и селективного разделения ионов вы знаете?
14. Вычислите 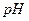 раствора серной кислоты с молярной концентрацией эквивалента 0,005 моль/дм3 (считая кислоту сильной по обеим степеням диссоциации).
раствора серной кислоты с молярной концентрацией эквивалента 0,005 моль/дм3 (считая кислоту сильной по обеим степеням диссоциации).
15. Рассчитайте молярную концентрацию ионов 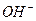 в растворе с
в растворе с 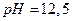 .
.
16. Вычислите раствора, содержащего в 1 дм3 0,2 г гидроксида натрия.
17. Рассчитайте 0,1 моль/дм3 раствора уксусной кислоты.
18. Рассчитайте 0,01 моль/дм3 раствора гидроксида аммония.
19. Рассчитайте константу гидролиза, степень гидролиза и 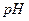 0,1 моль/дм3 раствора роданида аммония. Какие факторы усиливают и какие подавляют гидролиз?
0,1 моль/дм3 раствора роданида аммония. Какие факторы усиливают и какие подавляют гидролиз?
20. Вычислите ацетатной буферной смеси, содержащей по 0,1 моль/дм3 уксусной кислоты и ацетата натрия. Для чего применяются буферные растворы? Что такое буферная емкость?
21. Как изменится аммонийной буферной смеси, содержащей по 0,1 моль в 1 дм3 гидроксида и хлорида аммония, при добавлении к 1 дм3 смеси 0,01 моль соляной кислоты.
22. Вычислите произведение растворимости гидроксида магния, если его растворимость в 1 дм3 воды равна 0,0120 г.
23. Вычислите растворимость хлорида свинца в воде, если ПР = 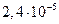 .
.
24. Выпадет ли осадок, если смешать 30 см 0,003 моль/дм3 раствора хромата калия и 20 см3 0,0002 моль/дм3 раствора нитрата серебра? (ПР = 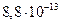 ).
).
2. Систематический анализ катионов (сульфидный метод)
Цель и задачи работы
Изучение принципов деления катионов на аналитические группы. Овладение навыками проведения групповых реакций и реакций обнаружения отдельных катионов, выполнения операций фильтрования, центрифугирования, декантации и др. Решение контрольной задачи — установление качественного состава смеси катионов I и II аналитических групп.
Программа работы
Обнаруживаемый катион
Вывод
2.3. Вопросы для самоподготовки
1. Чем отличается дробный метод анализа от систематического?
2. Какие реакции называются общими, специфическими, частными?
3. В чем преимущества систематического метода анализа смеси катионов?
4. Каковы преимущества дробного метода анализа, для решения каких задач его применяют?
5. На чем основано деление катионов на аналитические группы?
6. Какова связь деления катионов на аналитические группы с периодическим законом Менделеева?
7. Что называется внешним эффектом в качественном анализе?
8. Что такое маскировка иона и как она проводится?
9. Какие классификации ионов существуют в качественном анализе?
10. Какие требования предъявляются к реакциям в качественном анализе?
11. Что называется групповым реагентом?
12. Какие требования предъявляются к групповому реагенту?
13. Чем отличаются катионы I аналитической группы по сероводородному способу классификации катионов от других аналитических групп?
14. С чего начинается качественный систематический анализ?
15. Когда обнаруживают катионы I аналитической группы?
16. Почему в качестве группового реагента на катионы II группы применяют карбонат аммония, а не другие соединения (сульфат аммония, карбонат натрия, фосфат аммония)?
17. Чем обусловлено осаждение групповым реагентом катионов II группы при 800о С?
18. Как окрашены растворы солей катионов I и II групп?
19. Какую реакцию имеют водные растворы ацетатов катионов I и II групп?
20. Почему осаждение катионов II группы проводят при 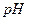 9?
9?
21. Почему осаждение катиона кальция проводят при 4-5?
22. Когда и как проводится определение иона аммония?
23. Почему влажная лакмусовая бумага изменяет окраску при обнаружении иона аммония в газовой камере?
24. Почему при обнаружении иона бария дихроматом калия в осадок выпадает хромат бария, а не дихромат?
25. Почему при осаждении иона бария добавляют избыток ацетата натрия?
26. Почему обнаружение иона калия проводят в отсутствие иона аммония?
27. Как устанавливают полноту осаждения?
28. Почему карбонаты катионов II аналитической группы растворяют в уксусной кислоте, а не в соляной?
29. Можно ли обнаружить ион кальция действием оксалата аммония в присутствии иона бария?
30. Перед выполнением лабораторных работ студент должен изучить инструкции по безопасным методам работы и расписаться в журнале регистрации.
Цель и задачи работы
Усвоение принципов гравиметрического анализа, его теории и практики. Отработка навыков проведения определений элементов и веществ, технологии выполнения отдельных операций: осаждение, декантация, фильтрование, сушка, прокаливание, взвешивание и др. Изучение условий применения различных реагентов — осадителей. Выполнение контрольного определения (анализ раствора вещества неизвестной концентрации).
Программа работы
Приборы и реактивы.
1. Муфельная печь;
2. Технохимические весы;
3. Аналитические весы;
4. Водяная баня;
5. Эксикатор;
6. Стеклянные палочки;
7. Стеклянные воронки;
8. Фарфоровые тигли;
9. Стеклянные фильтры № 3-20;
10. Щипцы тигельные;
11. Термостат;
12. Химические стаканы вместимостью 100, 200 и 250 см3;
13. Мерный цилиндр вместимостью 100 см3;
14. Пипетки;
15. Колба Бунзена;
16. Водоструйный или вакуумный насос;
17. Беззольные фильтры «синяя лента», «красная лента»;
18. Хлорид бария, водный раствор, С = 3 моль/дм3;
19. Хлороводородная кислота, водный раствор, С = 6 моль/дм3 и С = 2 моль/дм3;
20. Гидроксид аммония, водный раствор, w = 10% (масс.) и С = 2 моль/дм3;
21. Нитрат аммония, водный раствор, w = 2% (масс);
22. Азотная кислота, концентрированная;
23. Диметилглиоксим динатриевая соль, водный раствор, w = 1% (масс);
24. Йод, водно-спиртовой раствор, С = 0.025 моль/дм3;
25. Карбонат натрия, водный раствор,С = 1 моль/дм3;
26. Уксусная кислота концентрированная и водный раствор, w = 20% (масс), w = 5% (масс);
27. Пероксид водорода, водный раствор, w = 30% (масс);
28. Гидроксид натрия, водный раствор, С = 2 моль/дм3;
29. Нитрон, раствор в 5%-ной уксусной кислоте, С = 0,1 моль/дм3;
30. a-Нитрозо-b-нафтола (свежеприготовленный раствор). Растворяют 8 г a-нитрозо-b-нафтола в 300 см3 концентрированной уксусной кислоты и разбавляют до 500 см3 дистиллированной водой.
Определение сульфатов
Основано на осаждении сульфат-ионов в виде кристаллического белого осадка 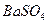 и получении гравиметрической формы
и получении гравиметрической формы 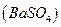 прокаливанием:
прокаливанием:
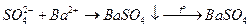
Данным методом определяют сульфатную серу, а также свободную, пиритную и сульфидную, предварительно окислив их до сульфатной.
Ход работы.
Навеску сульфата (или аликвотную часть анализируемого раствора) переносят в химический стакан вместимостью 200-250 см3 и приливают 100 см3 дистиллированной воды. К полученному раствору добавляют 2 см3 6 М раствора 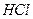 и нагревают почти до кипения на газовой горелке. В химический стакан вместимостью 100 см3 помещают 50 см3 дистиллированной воды и 6 см3 3 М раствора
и нагревают почти до кипения на газовой горелке. В химический стакан вместимостью 100 см3 помещают 50 см3 дистиллированной воды и 6 см3 3 М раствора 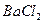 , нагревают почти до кипения на газовой горелке. Затем осторожно, по каплям, при постоянном перемешивании стеклянной палочкой, приливают нагретый раствор хлорида бария к горячему раствору сульфата, при этом палочка не должна касаться стенок стакана. Стакан с образовавшимся осадком ставят на кипящую водяную баню. Как только осадок осядет и жидкость над ним просветлеет, проводят проверку полноты осаждения сульфата бария. Для этого к раствору над осадком приливают 2-3 капли горячего раствора
, нагревают почти до кипения на газовой горелке. Затем осторожно, по каплям, при постоянном перемешивании стеклянной палочкой, приливают нагретый раствор хлорида бария к горячему раствору сульфата, при этом палочка не должна касаться стенок стакана. Стакан с образовавшимся осадком ставят на кипящую водяную баню. Как только осадок осядет и жидкость над ним просветлеет, проводят проверку полноты осаждения сульфата бария. Для этого к раствору над осадком приливают 2-3 капли горячего раствора 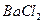 осторожно, не взмучивая осадка. Если при этом не наблюдается помутнения раствора, полнота осаждения достигнута, в противном случае приливают еще 1 см3 дают осадку осесть и снова проверяют полноту осаждения.
осторожно, не взмучивая осадка. Если при этом не наблюдается помутнения раствора, полнота осаждения достигнута, в противном случае приливают еще 1 см3 дают осадку осесть и снова проверяют полноту осаждения.
Затем стакан ставят на кипящую водяную баню на 2-3 часа для созревания осадка, после чего осадок фильтруют и промывают. Для фильтрования применяют бумажный беззольный фильтр «синяя лента». Промывная жидкость — дистиллированная вода, подкисленная 6 М раствором 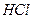 (8-10 капель на 100 см3 воды). Подсушивание, озоление фильтра и прокаливание осадка (600-800° С).
(8-10 капель на 100 см3 воды). Подсушивание, озоление фильтра и прокаливание осадка (600-800° С).
Осаждению сульфат-ионов в виде 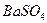 мешают анионы
мешают анионы 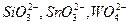 и другие, образующие осадки соответствующих кислот при подкислении раствора. Осаждению мешают также ионы
и другие, образующие осадки соответствующих кислот при подкислении раствора. Осаждению мешают также ионы 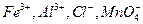 и другие, соосаждаемые с
и другие, соосаждаемые с  . Мешающие ионы должны быть предварительно удалены из анализируемого раствора.
. Мешающие ионы должны быть предварительно удалены из анализируемого раствора.
Массу сульфат-ионов в пробе рассчитывают по формуле
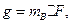
где 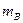 – масса гравиметрической (весовой) формы, г;
– масса гравиметрической (весовой) формы, г;
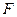 – гравиметрический фактор.
– гравиметрический фактор.
Содержание отделяемого вещества (г/дм3, моль/дм3, % масс и другие) устанавливают расчетом в соответствующей концентрации.
Результаты трех параллельных определений оформляют в таблицу 3.1 и делают их математическую обработку.
Результаты гравиметрического определения сульфатов.
Таблица 3.1.
| № опыта | Масса, г | Содержание (в соответствующих единицах) | |||
| Тигель или стеклянный фильтр | Гравиметрическая форма | Определяемое вещество | |||
| С гравиметрической формой | Пустой | ||||
Определение железа
Основано на осаждении железа (III) в виде аморфного, коричневого осадка 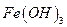 и получении гравиметрической формы
и получении гравиметрической формы 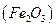 прокаливанием:
прокаливанием:
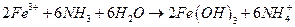
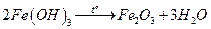 .
.
Ход работы.
Навеску образца, содержащего 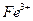 (или аликвотную часть анализируемого раствора), переносят в стакан вместимостью 100 см3, подкисляют 2-3 каплями концентрированной
(или аликвотную часть анализируемого раствора), переносят в стакан вместимостью 100 см3, подкисляют 2-3 каплями концентрированной 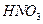 для окисления
для окисления 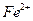 -ионов, возможно содержащихся в растворе, и для предотвращения гидролиза
-ионов, возможно содержащихся в растворе, и для предотвращения гидролиза 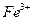 и нагревают на кипящей водяной бане до 70-800 С. К горячему раствору при постоянном перемешивании приливают 10%-й водный раствор аммиака до появления устойчивого запаха аммиака, после чего к раствору с осадком приливают 100 см3 горячей дистиллированной воды и ставят на кипящую водяную баню. После осаждения осадка, его фильтруют через бумажный беззольный фильтр «красная лента».
и нагревают на кипящей водяной бане до 70-800 С. К горячему раствору при постоянном перемешивании приливают 10%-й водный раствор аммиака до появления устойчивого запаха аммиака, после чего к раствору с осадком приливают 100 см3 горячей дистиллированной воды и ставят на кипящую водяную баню. После осаждения осадка, его фильтруют через бумажный беззольный фильтр «красная лента».
Осадок на фильтре промывают горячим 2%-м раствором нитрата аммония. Промывание заканчивают, когда проба на полноту промывания дает отрицательную реакцию на анион (например, при анализе хлорида железа (III) проба должна дать отрицательный результат на содержание 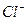 -ионов при добавлении к капле фильтрата капли и капли
-ионов при добавлении к капле фильтрата капли и капли 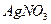 ). Промытый и подсушенный осадок озоляют и прокаливают в тигле при 1000—12000 С.
). Промытый и подсушенный осадок озоляют и прокаливают в тигле при 1000—12000 С.
Осаждению мешают катионы, осаждаемые водным раствором аммиака в виде гидроксидов 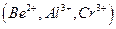 и др., а также анионы, которые в аммиачной среде образуют осадки с катионами, присутствующими в анализируемом растворе
и др., а также анионы, которые в аммиачной среде образуют осадки с катионами, присутствующими в анализируемом растворе 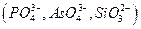 и др., осаждению мешают также катионы, соосаждаемые
и др., осаждению мешают также катионы, соосаждаемые 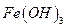 , и ионы, образующие комплексные соединения с
, и ионы, образующие комплексные соединения с 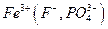 и др.. Все эти мешающие ионы перед проведением анализа должны быть удалены.
и др.. Все эти мешающие ионы перед проведением анализа должны быть удалены.
Расчет и оформление результатов анализа см. п. 3.2.1.
Определение никеля
Диметилглиоксим с ионами никеля (II) образует в аммиачной среде малорастворимый осадок диметилглиoксимата никеля ярко-розового цвета:
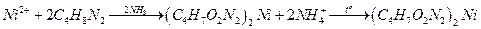
Ход работы.
Раствор, содержащий не более 30 мг никеля, разбавляют водой до 100 мл, подкисляют 2 М раствором 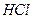 до слабокислой реакции, нагревают до 80°С и добавляют раствор реагента в небольшом избытке. Необходимо добавить 5 см3 реактива приблизительно на каждые 10 мг никеля, однако это соотношение должно быть увеличено, если в растворе присутствуют
до слабокислой реакции, нагревают до 80°С и добавляют раствор реагента в небольшом избытке. Необходимо добавить 5 см3 реактива приблизительно на каждые 10 мг никеля, однако это соотношение должно быть увеличено, если в растворе присутствуют 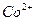 и
и 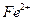 , образующие в этих условиях устойчивые хелаты. Затем добавляют 2 М раствор аммиака до появления запаха и продолжают нагревать на водяной бане не более 1 ч. Не следует проводить длительного нагревания при доступе воздуха, так как в этих условиях может образоваться нерастворимый хелат никеля. После проверки полноты осаждения, осадок пропускают через стеклянный фильтр и промывают теплой водой до исчезновения ионов
, образующие в этих условиях устойчивые хелаты. Затем добавляют 2 М раствор аммиака до появления запаха и продолжают нагревать на водяной бане не более 1 ч. Не следует проводить длительного нагревания при доступе воздуха, так как в этих условиях может образоваться нерастворимый хелат никеля. После проверки полноты осаждения, осадок пропускают через стеклянный фильтр и промывают теплой водой до исчезновения ионов 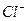 в промывных водах. Стеклянный фильтр с осадком сушат в термостате при 110-120° С до постоянной массы и взвешивают.
в промывных водах. Стеклянный фильтр с осадком сушат в термостате при 110-120° С до постоянной массы и взвешивают.
Расчет и оформление результатов анализа см. п. 3.2.1.
Определение кобальта
Определение основано на осаждении 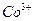 уксуснокислым раствором a-нитрозо-b-нафтола. Предварительно
уксуснокислым раствором a-нитрозо-b-нафтола. Предварительно 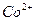 окисляют до . Состав полученного осадка соответствует формуле:
окисляют до . Состав полученного осадка соответствует формуле:
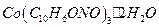
Раствор a-нитрозо- b-нафтола. Растворяют 8 г a - нитрозо - b - нафтола в 300 мл концентрированной уксусной кислоты и разбавляют до 500 см3 дистиллированной водой. Раствор готовят непосредственно перед употреблением.
Ход работы
К анализируемому раствору, содержащему не более 5 мг 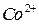 добавляют 20 капель 30%-ного раствора
добавляют 20 капель 30%-ного раствора 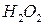 и 2 М раствор
и 2 М раствор 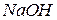 при перемешивании до начала образования черного осадка гидроксида
при перемешивании до начала образования черного осадка гидроксида 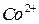 . Затем приливают 20 см3 концентрированной уксусной кислоты и, если осадок не растворился, раствор нагревают до его растворения. Разбавляют раствор до 100см3 раствора a-нитрозо-b-нафтола, доводят до кипения и продолжают нагревать на водяной бане 20-30 мин., при этом выпадает осадок красно-бурого цвета. Затем осадок фильтруют через стеклянный фильтр (№3-20 – 40 пор), промывают небольшими порциями горячего раствора уксусной кислоты и воды. Фильтр с осадком сушат при 130оС в сушильном шкафу до постоянной массы и взвешивают в виде
. Затем приливают 20 см3 концентрированной уксусной кислоты и, если осадок не растворился, раствор нагревают до его растворения. Разбавляют раствор до 100см3 раствора a-нитрозо-b-нафтола, доводят до кипения и продолжают нагревать на водяной бане 20-30 мин., при этом выпадает осадок красно-бурого цвета. Затем осадок фильтруют через стеклянный фильтр (№3-20 – 40 пор), промывают небольшими порциями горячего раствора уксусной кислоты и воды. Фильтр с осадком сушат при 130оС в сушильном шкафу до постоянной массы и взвешивают в виде 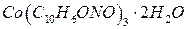 .
.
Расчет и оформление результатов анализа см. п. 3.2.1.
Определение перхлоратов
Нитрон с перхлорат-ионами в уксуснокислой среде образует малорастворимый осадок перхлората нитрония серовато-белого цвета:
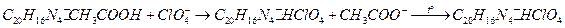
Ход работы.
Раствор, содержащий не более 100 мг перхлората, разбавляют водой до 50 см3, приливают 1 см3 ледяной уксусной кислоты, нагревают до 70° С и добавляют при перемешивании раствор нитрона в небольшом избытке. Необходимо добавить 1,5 см3 раствора на каждые 10 мг перхлората. Раствор охлаждают (охлаждение способствует полному выпадению осадка). Через 1 ч проверяют полноту осаждения. Осадок пропускают через стеклянный фильтр и промывают 2-3 раза 5% уксусной кислотой и 2 раза холодной дистиллированной водой. Тигель с осадком сушат при 100° С в термостате до постоянной массы и взвешивают.
Нитраты можно определять аналогично.
Расчет и оформление результатов анализа см. п. 3.2.1.
Протолитометрический анализ
Цель и задачи работы
Закрепление знаний теории протолитических равновесий и титриметрического анализа с применением кислотно-основных реакций. Получение навыков титрования с применением 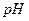 – индикоторов, проведения расчетов результатов анализа и их обработка. Контрольная задача – определение протолитов и в растворах протолитов и их смесей неизвестной концентрации.
– индикоторов, проведения расчетов результатов анализа и их обработка. Контрольная задача – определение протолитов и в растворах протолитов и их смесей неизвестной концентрации.
Программа работы
Перманганатометрия.
Цель и задачи работы.
Закрепление знаний теории окислительно-восстановительных равновесий и титриметрического анализа с применением окислительно-восстановительных реакций. Получение навыков титрования и проведения расчетов результатов анализа и их обработка. Освоение способов прямого, заместительного титрования и титрования по остатку. Контрольная задача — определение восстановителей в растворах неизвестной концентрации.
Программа работы
Определение железа (II).
Оборудование и реактивы:
1. Бюретка вместимостью 25 см3;
2. Мерные колбы вместимостью 100, 250 и 500 см3 ;
3. Колбы для титрования вместимостью 250 см3 ;
4. Пипетки Мора на 5, 10, 15, 25 см3;
5. Аналитические весы с разновесами;
6. Набор ареометров;
7. Щавелевая кислота 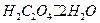 , х.ч. или ч.д.а.;
, х.ч. или ч.д.а.;
8. Перманганат калия 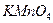 , х.ч. или ч.д.а.;
, х.ч. или ч.д.а.;
9. Серная кислота, раствор 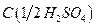 = 2 моль/дм3;
= 2 моль/дм3;
10. Фосфорная кислота, раствор 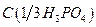 = 6 моль/дм3;
= 6 моль/дм3;
11. Соль Мора, раствор 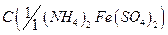 = 0,5 моль/дм3;
= 0,5 моль/дм3;
12. Хлороводородная кислота, раствор 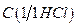 = 1
= 1 
 ;
;
13. Оксалат аммония, раствор 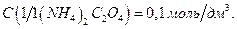 ;
;
14. Индикатор метиловый оранжевый, раствор 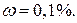 ;
;
15. Нитрат серебра, водный раствор, w = 1%;
16. Азотная кислота, 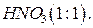 ;
;
17. Хлорид кальция, раствор 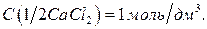
5.2.1.1. Приготовление первичного стандарта щавелевой кислоты 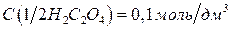
Выполняют по п.4.2.1.1. из реактива квалификации х.ч. учитывая, что 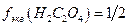 . Раствор следует готовить примерно такой же концентрации, как раствор
. Раствор следует готовить примерно такой же концентрации, как раствор 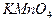 .
.
5.2.1.2. Приготовление и стандартизация рабочего раствора перманганата калия (вторичного стандарта) 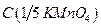 =0,1 моль/дм3
=0,1 моль/дм3
Применяемый для приготовления растворов титранта твердый перманганат калия всегда в какой-то степени загрязнён диоксидом марганца, катализирующим процесс разложения перманганата в растворе. Удаление диоксида марганца фильтрованием заметно повышает устойчивость растворов перманганата. Фильтрование следует проводить спустя некоторое время после приготовления раствора, чтобы произошло полное окисление примесей в воде, для ускорения реакции окисления раствор можно прокипятить. Для фильтрования нельзя применять бумажный фильтр, так как целлюлоза реагирует с перманганатом, образуя диоксид марганца.
Разложение раствора ускоряется на свету, при нагревании, под действием кислот, оснований, ионов 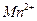 и диоксида марганца:
и диоксида марганца:
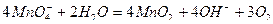
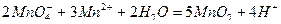
Стандартные растворы 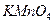 хранят в темном месте в темных склянках со стеклянными пробками.
хранят в темном месте в темных склянках со стеклянными пробками.
Для установки характеристик раствора перманганата обычно используют свежеперекристаллизованную щавелевую кислоту 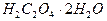 , оксалат натрия
, оксалат натрия 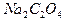 или оксалат аммония
или оксалат аммония 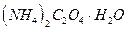 . Реакцию между перманганат- и оксалат-ионами в кислой среде можно записать:
. Реакцию между перманганат- и оксалат-ионами в кислой среде можно записать:
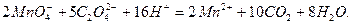
Ход работы:
1. Рассчитывают навеску, учитывая, что 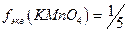 (кислая среда);
(кислая среда);
2. На технических весах в стеклянном бюксе взвешивают рассчитанную навеску исходного перманганата.
3. Количественно переносят навеску дистиллированной водой в мерную колбу, растворяют при перемешивании и доводят раствор до метки дистиллированной водой.
4. Свежеприготовленный раствор переносят в стакан, нагревают до кипения и поддерживают эту температуру приблизительно 1 ч. Затем раствор охлаждают и фильтруют через стеклянный, пористый фильтр. Можно перед фильтрованием оставить раствор при комнатной температуре на несколько дней.
5. Аликвотную часть раствора щавелевой кислоты 10 см3 переносят пипеткой в коническую колбу для титрования, добавляют 10 см3 раствора 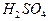 и нагревают на водяной бане примерно до 75-800С. В начале титрования следует приливать из бюретки раствор к горячему раствору оксалата по каплям. Каждую следующую каплю прибавляют после того, как исчезнет окраска от предыдущей. Титрование заканчивают, когда избыточная капля раствора окрасит раствор в бледно-розовый цвет, не исчезающий 1 мин.
и нагревают на водяной бане примерно до 75-800С. В начале титрования следует приливать из бюретки раствор к горячему раствору оксалата по каплям. Каждую следующую каплю прибавляют после того, как исчезнет окраска от предыдущей. Титрование заканчивают, когда избыточная капля раствора окрасит раствор в бледно-розовый цвет, не исчезающий 1 мин.
6. Проводят 3-5 титрований. Результаты заносят в таблицу 5.1:
Результаты стандартизации раствора перманганата калия.
Таблица 5.1
| № п/п | V(Н2С2О 4), см3 | С(!/2Н2С2О4 ), моль/дм3 | V(KMnO4), см3 | С(1/5KMnO4), моль/дм3 | Т(KMnO4), г/см3 |
Используя средний объем титранта, затраченного на титрование, рассчитывают характеристики раствора 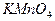 . При расчете учитывают, что в точке эквивалентности число молей эквивалента титруемого вещества и титранта равны:
. При расчете учитывают, что в точке эквивалентности число молей эквивалента титруемого вещества и титранта равны: 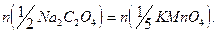
Определение кальция
Ионы 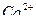 определяют косвенно. Для этого ионы осаждают раствором оксалата аммония, взятом в избытке:
определяют косвенно. Для этого ионы осаждают раствором оксалата аммония, взятом в избытке:
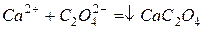
Осадок отфильтровывают, промывают дистиллированной водой и затем растворяют на фильтре в разбавленной серной кислоте (ПР 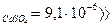 ПР
ПР 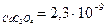
 ), при этом образуется эквивалентное количество щавелевой кислоты, которую титруют стандартным раствором перманганата калия:
), при этом образуется эквивалентное количество щавелевой кислоты, которую титруют стандартным раствором перманганата калия:
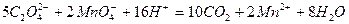
Ход работы:
1. Исследуемый раствор, содержащий , в мерной колбе разбавляют дистиллированной водой до метки и тщательно перемешивают.
2. Аликвотную часть анализируемого раствора переносят пипеткой в химический стакан вместимостью 200—250 см3, добавляют 100 см3 дистиллированной воды, 1—2 см3 раствора 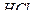 , 1—2 капли индикатора метилового оранжевого и нагревают до 70—80°С.
, 1—2 капли индикатора метилового оранжевого и нагревают до 70—80°С.
3. Медленно, по каплям (1—2 капли в 1 секунду), при непрерывном перемешивании добавляют 1,5-кратный избыток нагретого раствора оксалата аммония.
4. После добавления раствора осадителя прибавляют по каплям при перемешивании разбавленный раствор аммиака до исчезновения розовой окраски раствора и оставляют раствор с осадком на 1 часа на кипящей водяной бане для созревания осадка. Параллельно проводят осаждение не менее трех проб.
5. Созревший осадок оксалата кальция количественно переносят на плотный фильтр и тщательно промывают несколько раз небольшими порциями разбавленного раствора оксалата аммония для удаления ионов 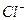 . Признаком конца промывания может служить отсутствие белого осадка при действии на порцию (2—3 капли) промывных вод, подкисленную
. Признаком конца промывания может служить отсутствие белого осадка при действии на порцию (2—3 капли) промывных вод, подкисленную 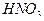 , раствором
, раствором 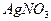 .
.
6. Затем промывают осадок дистиллированной водой для удаления избытка оксалат-ионов (отсутствие белого осадка при действии раствора на 2—3 капли промывных вод). Большого избытка промывных вод следует избегать, так как это может вызвать заметные потери за счет растворения осадка оксалата кальция.
7. Бумажный фильтр прокалывают в центре острием стеклянной палочки, которую обмывают над фильтром серной кислотой, и тщательно промывают его 40—50 см3 раствора и 2 раза дистиллированной водой порциями по 10 см3.
Полученный раствор 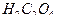 нагревают на водяной бане до 70— 80°С и титруют стандартным раствором до появления бледно-розовой окраски, не исчезающей 1 минуту. Результаты заносят в таблицу 5.3:
нагревают на водяной бане до 70— 80°С и титруют стандартным раствором до появления бледно-розовой окраски, не исчезающей 1 минуту. Результаты заносят в таблицу 5.3:
Результаты титрования раствора щавелевой кислоты
раствором перманганата калия.
Таблица 5.3.
| № п/п | V(Н2С2О 4), см3 | С(1/2Н2С2О4 ), моль/дм3 | V(KMnO4), см3 | С(1/5KМnO4), моль/дм3 | Т(KMnO4), г/см3 |
8. Расчет содержания проводят с учетом того, что их содержание эквивалентно содержанию оксалат-ионов. В точке эквивалентности
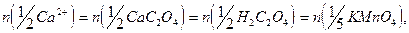
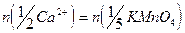
Содержание (в г) рассчитывают по формулам:
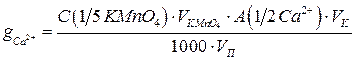
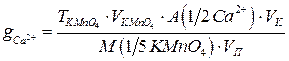
5.3. Вопросы для самоподготовки.
1. Чем определяется величина скачка на кривой титрования? Приведите примеры приёмов увеличения скачка титрования.
2. В каких случаях кривая титрования симметрична, а в каких — асимметрична относительно точки эквивалентности? Приведите примеры.
3. Каковы особенности приготовления стандартного раствора и условия его хранения?
4. Какими химическими реакциями, протекающими в растворе, обуславливается изменение концентрации перманганата калия?
5. Почему фактор эквивалентности различен при проведении реакций в кислой, щелочной или нейтральной средах? Рассчитайте его значение в этих средах.
6. Какие вещества применяют для установки характеристик раствора ? Какие требования к ним предъявляют?
7. Назовите первичные стандарты, применяемые для установления концентрации перманганата калия.
8. Напишите уравнение реакции взаимодействия перманганат-иона с оксалат-ионом.
9. Как определить перманганатометрическим методом вещества, не обладающие окислительно-восстановительными свойствами, например 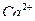 ?
?
10. Как определить окислители перманганатометрическим методом: 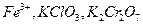 ?
?
11. Назовите окислители и восстановители, применяемые для предварительного окисления и восстановления титруемых веществ.
12. Какие восстановители применяют для предварительного восстановления железа (III)? Напишите реакции.
13. Объясните роль защитных смесей при титровании железа (II) перманганатом калия в присутствии хлорид-иона.
14. Назовите компоненты смеси Рейнгарда-Циммермана и объясните их роль в процессе титрования железа (II).
15. В каких условиях проводят определение ионов 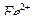 при отсутствии хлорид-ионов перманганатометрическим методом?
при отсутствии хлорид-ионов перманганатометрическим методом?
16. Вычислите молярную концентрацию эквивалентов раствора с Т= 0,005815 г/см3 при использовании его в качестве титра в кислой и нейтральной средах.
17. В мерной колбе вместимостью 100 см3 приготовлен раствор, содержащий 0,6384 г 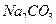 . Какой объём 0,09349
. Какой объём 0,09349 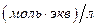 раствора будет израсходован на титрование 10,00 см3 раствора оксалата натрия? Рассчитайте константу равновесия окислительно-восстановительной реакции, пользуясь таблицей окислительно-восстановительных потенциалов.
раствора будет израсходован на титрование 10,00 см3 раствора оксалата натрия? Рассчитайте константу равновесия окислительно-восстановительной реакции, пользуясь таблицей окислительно-восстановительных потенциалов.
18. На титрование раствора 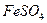 израсходовано 17,25 см3 раствора с
израсходовано 17,25 см3 раствора с 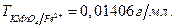 . Сколько граммов железа содержится в пробе?
. Сколько граммов железа содержится в пробе?
Иодометрия
Цель и задачи работы.
Закрепление знаний теории окислительно-восстановительного равновесия и титриметрического анализа с применением окислительно-восстановительных реакций. Получение навыков титрования и проведения расчетов результатов анализа и их обработки. Контрольная задача – определение окислителей в растворах неизвестной концентрации.
Оборудование и реактивы:
1. Бюретка вместимостью 25 см3;
2. Мерные колбы вместимостью 00, 250 и 500 см3 ;
3. Колбы для титрования вместимостью 250 см3 ;
4. Пипетки Мора на 5, 10, 15, 25 см3;
5. Аналитические весы с разновесами;
6. Набор ареометров;
7. Бихромат калия 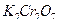 , х.ч. или ч.д.а.;
, х.ч. или ч.д.а.;
8. Тиосульфат натрия 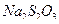 , х.ч. или ч.д.а.;
, х.ч. или ч.д.а.;
9. Иодид калия 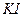 , х.ч. или ч.д.а.;
, х.ч. или ч.д.а.;
10. Серная кислота, раствор 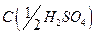 = 2 моль/дм3;
= 2 моль/дм3;
11. Сульфат меди (II), раствор 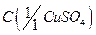 = 0,5 моль/дм3;
= 0,5 моль/дм3;
12. Крахмал, водный раствор.
Программа работы
Определение меди (II)
6.2.1.1. Приготовление первичного стандарта бихромата калия С(1/6K2Сr2О7 ) = 0,05 моль/дм3
Выполняют по п. 4.2.1.1. из реактива 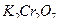 квалификации х.ч учитывая, что ¦экв.
квалификации х.ч учитывая, что ¦экв. 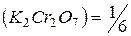 .
.
6.2.1.2. Приготовление рабочего раствора тиосульфата натрия С(1/1Na2S2О3) = 0,05 моль/дм3(вторичного стандарта)
Выполняют по п. 4.2.1.1. из реактива 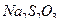 квалификации х.ч учитывая, что ¦экв.
квалификации х.ч учитывая, что ¦экв. 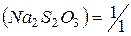 . Для приготовления растворов рекомендуется свежепрокипяченная (в течение часа) вода, так как бактерии разлагают растворы тиосульфата. Для подавления роста бактерий можно добавлять такие вещества, как хлороформ, бензоат натрия или
. Для приготовления растворов рекомендуется свежепрокипяченная (в течение часа) вода, так как бактерии разлагают растворы тиосульфата. Для подавления роста бактерий можно добавлять такие вещества, как хлороформ, бензоат натрия или 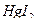 . Кипячение воды и добавление небольших количеств (0,1 г/ дм3) способствует удалению
. Кипячение воды и добавление небольших количеств (0,1 г/ дм3) способствует удалению 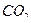 , влияющего на характеристики раствора. Раствор хранят в темном месте в хорошо закрытой посуде. Стандартизацию раствора проводят обычно через 5—7 дней.
, влияющего на характеристики раствора. Раствор хранят в темном месте в хорошо закрытой посуде. Стандартизацию раствора проводят обычно через 5—7 дней.
6.2.1.3. Стандартизация рабочего раствора тиосульфата натрия (вторичного стандарта).
Важнейшими факторами, определяющими устойчивость раствора тиосульфата, являются значения 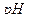 , присутствие микроорганизмов, концентрация раствора, присутствие атмосферного кислорода, примесей ионов тяжелых металлов (катализаторов окисления тиосульфата кислородом воздуха), воздействие прямого солнечного света.
, присутствие микроорганизмов, концентрация раствора, присутствие атмосферного кислорода, примесей ионов тяжелых металлов (катализаторов окисления тиосульфата кислородом воздуха), воздействие прямого солнечного света.
Первичными стандартами для растворов могут быть окислители: 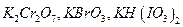 , выделяющие при взаимодействии с избытком иодид-ионов эквивалентное количество иода, который титруют стандартизируемым раствором тиосульфата. Способ стандартизации бихроматом калия основан на реакциях:
, выделяющие при взаимодействии с избытком иодид-ионов эквивалентное количество иода, который титруют стандартизируемым раствором тиосульфата. Способ стандартизации бихроматом калия основан на реакциях:
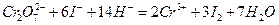
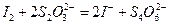
Ход работы:
1. В коническую колбу для титрования переносят пипеткой аликвотную часть раствора установочного вещества , добавляют 10 см3 раствора 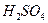 и 1 г кристаллического (или соответствующий объем его концентрированного раствора).
и 1 г кристаллического (или соответствующий объем его концентрированного раствора).
2. Колбы закрывают стеклянными пробками, содержимое перемешивают, дают постоять 5—10 мин в темном месте пока не завершится реакция.
3. Выделившийся 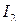 титруют раствором до тех пор, пока окраска не станет слабо-желтой.
титруют раствором до тех пор, пока окраска не станет слабо-желтой.
4. Затем титруемый раствор разбавляют водой приблизительно в 2 раза, добавляют индикатор крахмал и продолжают титрование до перехода окраски раствора из синей в светло-зеленую (ионы 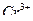 ).
).
5. Проводят 3-5 титрований. Результаты заносят в таблицу 6.1:
Результаты стандартизации раствора тиосульфата натрия.
Таблица 6.1.
| № п/п | V(K2Сr2О7), См3 | С(1/6K2Сr2О7), моль/дм3 | V(Na2S2О3), см3 | С(1/1Na2S2О3), моль/дм3 | Т(Na2S2О3), г/см3 |
Используя средний объем титранта, затраченного на титрование, рассчитывают характеристики раствора . При расчете учитывают, что в точке эквивалентности число молей эквивалента титруемого вещества и титранта равны: 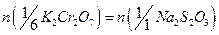 .
.
6.2.1.4. Контрольная задача: Определение меди (II).
Определение основано на взаимодействии ионов 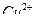 и
и 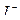 и последующем титровании образовавшегося в эквивалентном количестве
и последующем титровании образовавшегося в эквивалентном количестве 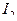 стандартным раствором
стандартным раствором 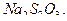
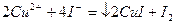
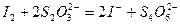
Ход определения:
1. Исследуемый раствор, содержащий ионы , в мерной колбе разбавляют дистиллированной водой до метки и тщательно перемешивают.
2. Аликвотные части анализируемого раствора пипеткой переносят в 3 конические колбы для титрования, добавляют по 2—3 г твердого  и 5 см3 раствора .
и 5 см3 раствора .
3. Колбы закрывают стеклянными пробками, которые перед титрованием обмывают дистиллированной водой и оставляют на 10-15 мин в темном месте.
4. Выделившийся  после завершения реакции титруют стандартным раствором
после завершения реакции титруют стандартным раствором 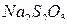 в присутствии крахмала.
в присутствии крахмала.
5. Результаты заносят в таблицу 6.2:
Результаты титрования раствора, содержащего ионы ,
раствором тиосульфата натрия.
Таблица 6.2.
| № п/п | V(Na2S2О3), см3 | С(1/1Na2S2О3), моль/дм3 | V(Cu2+), см3 | С(1/1 Cu2+), моль/дм3 | Т(Cu2+), г/см3 |
6. Используя средний объем титранта, израсходованного на титрование, рассчитывают содержание меди (II), учитывая, что в точке эквивалентности 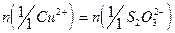 . Содержание (в г) вычисляют по формулам:
. Содержание (в г) вычисляют по формулам:
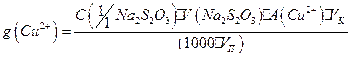
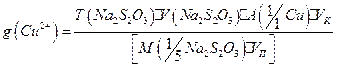
где 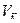 ,
, 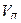 — вместимость колбы и пипетки соответственно.
— вместимость колбы и пипетки соответственно.
6.3. Вопросы для самоподготовки.
1. Почему иодид калия нельзя использовать для прямого титрования меди?
2. Напишите реакции, протекающие при титриметрическом определении:
а) белильной извести, нитратов, железа (III), пероксида водорода, брома, хлора, хрома (VI), хрома (III), сульфидов, сульфитов иодометрически; б) пероксида водорода, нитратов, диоксида марганца, кальция (II), марганца (II) перманганатометрически.
3. Каковы особенности приготовления стандартного раствора 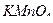 и условия его хранения?
и условия его хранения?
4. Почему фактор эквивалентности различен при проведении реакций в кислой, щелочной или нейтральной средах? Рассчитайте его значение в этих средах.
5. Какие вещества применяют для установки характеристик раствора 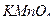 ? Какие требования к ним предъявляют?
? Какие требования к ним предъявляют?
6. Как определить перманганатометрическим методом вещества, не обладающие окислительно-восстановительными свойствами, например 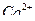 ?
?
7. Как определить окислители перманганатометрическим методом: 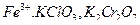 ?
?
8. Вычислите молярную концентрацию эквивалентов раствора 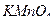 с Т= 0,005815 г/мл при использовании его в качестве титра в кислой и нейтральной средах.
с Т= 0,005815 г/мл при использовании его в качестве титра в кислой и нейтральной средах.
9. В каких условиях проводят определение ионов при отсутствии хлорид-ионов перманганатометрическим методом?
10. В мерной колбе вместимостью 100 мл приготовлен раствор, содержащий 0,6384 г . Какой объём 0,09349 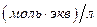 раствора будет израсходован на титрование 10,00 мл раствора оксалата натрия? Рассчитайте константу равновесия окислительно-восстановительной реакции, пользуясь таблицей окислительно-восстановительных потенциалов.
раствора будет израсходован на титрование 10,00 мл раствора оксалата натрия? Рассчитайте константу равновесия окислительно-восстановительной реакции, пользуясь таблицей окислительно-восстановительных потенциалов.
11. На титрование раствора израсходовано 17,25 мл раствора с Сколько граммов железа содержится в пробе?
12. Сравните достоинства и недостатки бихроматометрии с другими методами титриметрического анализа с использованием окислительно-восстано-вительных реакций.
13. Почему титрование растворов бихромата проводят в кислой среде?
14. Напишите окислительно-восстановительные реакции для дифениламина.
15. Рассчитайте погрешность титрования железа (II) бихроматом: а) в присутствии фосфорной кислоты ; б) в отсутствии фосфорной кислоты.
16. Как устанавливают конец титрования при броматометрическом титровании сурьмы (III)?
Комплексонометрия.
Цель и задачи работы.
Закрепление знаний теории реакций комплексообразования и комплексометрического титрования на примере комплексонометрии. Получение навыков комплексонометрического титрования, использования металлоиндикаторов, проведения расчетов результатов анализа и их обработки.
Контрольная задача — определение ионов металлов в растворах неизвестной концентрации.
Оборудование и реактивы:
1. Бюретка вместимостью 25 см3;
2. Мерные колбы вместимостью 100, 250 и 500 см3;
3. Колбы для титрования вместимостью 250 см3;
4. Пипетки Мора на 5, 10, 15, 25 см3;
5. Аналитические весы с разновесами;
6. Набор ареометров;
7. ЭДТА, раствор 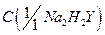 = 0,025 моль/дм3;
= 0,025 моль/дм3;
8. Хлорид кальция, раствор 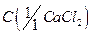 = 0,025 моль/дм3;
= 0,025 моль/дм3;
9. Хлорид железа (III), раствор 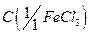 = 0,025 моль/дм3;
= 0,025 моль/дм3;
10. Аммиачный буферный раствор с 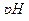 =10;
=10;
11. Ацетатный буферный раствор, = 4,5;
12. Комплексонат магния, 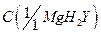 = 0,025 моль/дм3;
= 0,025 моль/дм3;
13. Эриохромовый черный Т, смесь с хлоридом натрия в отношении 1:100;
14. Сульфосалициловая кислота, раствор 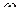 = 5%-й раствор.
= 5%-й раствор.
Программа работы
Определение кальция
Комплексонометрическое определение кальция основано на прямом титровании его раствором двунатриевой соли - этилендиаминтетрауксусной кислоты - 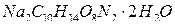 (ЭДТА) в присутствии мурексида, эриохромового черного Т или другого индикатора.
(ЭДТА) в присутствии мурексида, эриохромового черного Т или другого индикатора.
Реакция протекает по следующему уравнению:
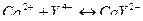
Образующийся хелат обладает невысокой устойчивостью, 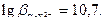 Ионы водорода, выделяющиеся в процессе титрования, приводят к смещению равновесия данной реакции в сторону разрушения комплекса.
Ионы водорода, выделяющиеся в процессе титрования, приводят к смещению равновесия данной реакции в сторону разрушения комплекса.
Оптимальным  для определения ионов кальция будет =11.
для определения ионов кальция будет =11.
При титровании с эриохромовым черным Т изменение окраски в конечной точке титрования недостаточно четкое, что можно объяснить незначительной устойчивостью образующегося металлоиндикаторного комплекса. Для получения более резкого изменения окраски индикатора в конечной точке титрования к анализируемому раствору перед титрованием можно добавить небольшое количество комплексоната магния. Переход окраски индикатора в конечной точке титрования становится очень четким. При этом протекает реакция:
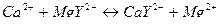
Исчезает окраска комплекса магния с эриохромовым черным Т, и появляется окраска свободного индикатора.
Ход работы:
1. Анализируемый раствор переносят в мерную колбу вместимостью 100 см3 доводят до метки дистиллированной водой и тщательно перемешивают.
2. В коническую колбу для титрования переносят аликвотную часть раствора (10 см3), добавляют 5 см3 аммиачного буферного раствора, 1 см3 раствора комплексоната магния, индикатора эриохромового черного Т на кончике шпателя и после полного растворения индикатора титруют раствором ЭДТА до изменения окраски индикатора от красно-фиолетовой к синей.
3. Титрование повторяют 3-5 раз до получения не менее трех сходящихся результатов титрования. Результаты заносят в таблицу 7.1:
Результаты титрования раствора кальция раствором ЭДТА.
Таблица 7.1.
| № п/п | V( Na2H2Y), см3 | С(1/1 Na2 H2 Y), моль/дм3 | V(Са) , см3 | С(1/1 Са), моль/дм3 | Т(Са), г/см3 |
4. Используя средний объем титранта, израсходованного на титрование, рассчитывают содержание кальция в растворе и количество определяемого вещества, учитывая что в точке эквивалентности образуется комплекс состава 1:1.
Определение железа
Определение железа (III) основано на прямом титровании его раствором ЭДТА в присутствии салициловой или сульфосалициловой кислот, тайрона, роданидов, хромазурола S, бензогидроксамовой кислоты. Реакция протекает по уравнению
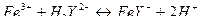
Железо (III) образует с ЭДТА один из самых устойчивых комплексов, 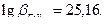 . Высокое значение константы устойчивости комплексоната железа позволяет проводить титрование в кислых средах при <3. Наиболее удобно проводить титрование при =2,5, при данной кислотности раствора логарифм условной константы устойчивости комплексоната железа равен
. Высокое значение константы устойчивости комплексоната железа позволяет проводить титрование в кислых средах при <3. Наиболее удобно проводить титрование при =2,5, при данной кислотности раствора логарифм условной константы устойчивости комплексоната железа равен 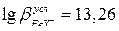 . Титрование железа возможно и при более низких значениях
. Титрование железа возможно и при более низких значениях 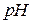 раствора, но при этом значительно возрастает коэффициент побочных реакций ЭДТА, что приводит к уменьшению условной константы устойчивости комплексоната железа, следовательно, к увеличению погрешности определения.
раствора, но при этом значительно возрастает коэффициент побочных реакций ЭДТА, что приводит к уменьшению условной константы устойчивости комплексоната железа, следовательно, к увеличению погрешности определения.
Железо (II) также образует с ЭДТА комплекс, но устойчивость этого комплекса ( 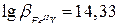 ) значительно меньше устойчивости комплексоната железа (III). При =2,5 железо (II) с ЭДТА не взаимодействует, поэтому перед титрованием его необходимо окислить до железа (III) нагреванием с
) значительно меньше устойчивости комплексоната железа (III). При =2,5 железо (II) с ЭДТА не взаимодействует, поэтому перед титрованием его необходимо окислить до железа (III) нагреванием с 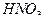 или добавлением пероксида водорода. Определение в кислых средах более избирательно. Определению железа (III) не мешают
или добавлением пероксида водорода. Определение в кислых средах более избирательно. Определению железа (III) не мешают 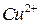 ,
, 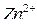 ,
, 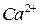 ,
, 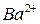 ,
, 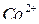 ,
, 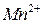 ,
, 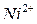 .
.
Ход работы
1. Анализируемый раствор в мерной колбе вместимостью 100 мл доводят до метки дистиллированной водой и тщательно перемешивают.
2. В коническую колбу для титрования переносят аликвотную часть раствора (10 см3), добавляют 5 см3 ацетатного буферного раствора, нагревают до 60 °С, добавляют 1 см3 раствора сульфосалициловой кислоты и титруют раствором ЭДТА до изменения окраски раствора от красно-фиолетовой к желтой.
3. Титрование повторяют 3-5 раз до получения не менее трех сходящихся результатов титрования. Результаты заносят в таблицу 7.2:
Результаты титрования раствора железа (III) раствором ЭДТА.
Таблица 7.2.
| № п/п | V( Na2H2Y), См3 | С(1/1 Na2H2Y), моль/дм3 | V(Fe(III), см3 | С(1/1 Fe(III), моль/дм3 | Т(Fe(III), г/см3 |
Используя средний объем титранта, израсходованного на титрование, рассчитывают содержание железа (III) в растворе и количество определяемого вещества, учитывая, что в точке эквивалентности образуется комплекс состава 1:1.
7.3. Вопросы для самоподготовки
1. Перечислите основные требования к реакциям, применяемым в методе комплексометрического титрования?
2. Какие факторы влияют на величину титрования?
3. Чем объясняется ограниченность применения неорганических реагентов в комплексометрическом титровании?
4. Назовите способы обнаружения конечной точки титрования.
5. Объясните сущность прямого, обратного, вытеснительного и косвенного приёмов комплексометрического титрования. В каких случаях применяется каждый из названных приёмов?
6. Что такое металлохромные индикаторы?
7. Напишите равновесие в растворе металлохромного индикатора.
8. Приведите графическую формулу ЭДТА. Какова дентатность ЭДТА?
9. Какова стехиометрия компонентов ЭДТА? Приведите графическую формулу комплексов двух- и трёхзарядных ионов металлов с ЭДТА.
10. Напишите уравнения реакций образования протонированных, нормальных и гидроксокомплексов ЭДТА.
11. Как готовят раствор ЭДТА?
12. Какие вещества могут быть использованы для стандартизации раствора ЭДТА?
13. В каких условиях проводят титрование ионов кальция раствором ЭДТА?
14. Как проводят комплексометрическое определение кальция и магния при совместном присутствии?
15. Почему следует удалить аммонийные соли при титровании кальция в присутствии магния?
16. В каких условиях проводят титрование железа (III) раствором ЭДТА? Укажите состав комплекса железа с сульфосалициловой кислотой в условиях проведения титрования.
17. Почему ионы трёхвалентных металлов титруют раствором ЭДТА в кислой среде, а щёлочноземельных — в щелочной?
18. Как можно предотвратить образование гидроксидов ионов металлов 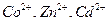 при их титровании раствором ЭДТА в щелочной среде?
при их титровании раствором ЭДТА в щелочной среде?
19. Предложите способ комплексометрического определения ионов 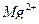 в присутствии
в присутствии 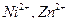 , ионов
, ионов 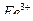 в присутствии
в присутствии 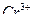 , ионов
, ионов 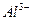 в присутствии
в присутствии 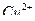 .
.
20. Рассчитайте навеску металлического цинка для установления характеристик ЭДТА методом отдельных навесок, чтобы на её титрование после растворения расходовалось 10,00 мл 0,0505 М раствора ЭДТА.
Ответ: 0,0330 г.
21. Из навески 1,200 г образца, содержащего хромат калия, приготовлен раствор в мерной колбе вместимостью 100 мл. К 25,00 мл полученного раствора прилит избыток раствора нитрата свинца. Полученный осадок отфильтровали, промыли, перевели в раствор и обработали 10,00 мл 0,1000 М раствора ЭДТА, избыток которого оттитровали 8,00 мл 0,05105 М раствора 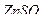 . Рассчитайте процентное содержание хромат-ионов в образце.
. Рассчитайте процентное содержание хромат-ионов в образце.
Ответ: 23,27%.
22. Вычислите процентное содержание 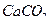 и
и 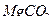 в известняке, если после растворения 1,0000 г пробы и соответствующей обработки раствора довели водой до 100 мл, на титрование 20 мл для определения суммы
в известняке, если после растворения 1,0000 г пробы и соответствующей обработки раствора довели водой до 100 мл, на титрование 20 мл для определения суммы 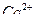 и затратили 19,25 мл 0,05140 М раствора ЭДТА, а на титрование израсходовали 6,26 мл раствора ЭДТА.
и затратили 19,25 мл 0,05140 М раствора ЭДТА, а на титрование израсходовали 6,26 мл раствора ЭДТА.
Ответ: — 16,0 %; — 28,1 %.
Инструментальные методы анализа
|
Цель и задачи работы
Изучение принципа и режимов работы пламенного фотометра, овладение навыками практического применения пламенно-фотометрического метода анализа, методикой построения градуировочных графиков и расчетами определения элементов градуировочных графиков, методом ограничивающих растворов, методом добавок и методом трёх эталонов.
Программа работы
Приборы и реактивы:
1. Пламенный фотометр - ПФМ;
2. Компрессор;
3. Рабочие растворы хлорида калия, хлорида натрия с концентрацией в пересчёте на калий, натрий 100 мкг/см3 и хлорида кальция с концентрацией в пересчёте на кальций 500 мкг/см3. Навески квалификации х.ч. 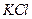 (0,019 г),
(0,019 г), 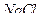 (0,0254 г) и
(0,0254 г) и 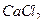 (0,1390 г) вносят в мерные колбы вместимостью 100 см3, доводят растворы до метки дистиллированной водой и тщательно перемешивают;
(0,1390 г) вносят в мерные колбы вместимостью 100 см3, доводят растворы до метки дистиллированной водой и тщательно перемешивают;
4. Мерные колбы вместимостью 100 см3, 7шт;
5. Градуированная пипетка вместимостью 10 см3, 1шт;
6. Мерные пробирки вместимостью 25 см3, 7шт.
Вопросы для самоконтроля
1. Какова природа происхождения атомных спектров?
2. На использовании какого явления основана пламенная фотометрия?
3. На измерении каких величин осуществляется качественный и количественный анализ в атомных спектральных методах?
4. Что определяется термином «потенциал возбуждения». Как он соотносится с интенсивностью спектральной линии?
5. Приведите эмпирическую зависимость интенсивности излучения от концентрации определяемого компонента.
6. Какие процессы происходят с веществами в пламени катализатора?
7. В чём заключается явление самопоглощения? Как оно влияет на проведение анализа?
8. Приведите принципиальную схему прибора для атомно-эмиссионной спектроскопии (АЭС).
9. Какие отличия имеет пламенный фотометр от приборов АЭС? Покажите на принципиальной схеме.
10. Каково назначение основных узлов пламенного фотометра?
11. Какие элементы и почему можно анализировать методами АЭС и пламенной фотометрии?
12. Какие преимущества и недостатки имеет пламенная фотометрия перед АЭС?
13. В чём заключаются методы определения элементов АЭС:
- градуировочного графика;
- трёх эталонов;
- добавок;
- ограничивающих растворов?
14. Каковы основные характеристики атомно-эмиссионного метода анализа?
Фотометрический анализ
Цель и задачи работы
Изучение работы фотометрических приборов, овладение навыками практического применения фотометрического метода анализа.
Программа работы
Выбор светофильтра
Раствор, имеющий наиболее интенсивную окраску, фотометрируют относительно раствора сравнения (воды), со всеми светофильтрами поочерёдно, записывая результаты измерений в таблицу 9.1.
Строят спектральную характеристику раствора в координатах 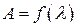 и выбирают светофильтр, соответствующий наибольшему значению светопоглощения.
и выбирают светофильтр, соответствующий наибольшему значению светопоглощения.
Данные для построения спектральной характеристики аммиаката меди (II).
Таблица 9.1
| № светофильтра | l, нм | Оптическая плотность |
Вопросы для самоконтроля
1. Какова природа возникновения спектров поглощения?
2. Спектры каких переходов используются в фотометрии?
3. Основной закон светопоглощения и его параметры?
4. Какие факторы влияют на оптическую плотность раствора и молярный коэффициент светопоглощения?
5. Физический смысл молярного коэффициента светопоглощения?
6. Причины отклонения от закона Бугера-Ламберта-Бера?
7. Классификация фотометрических методов анализа?
8. Принципиальная схема приборов для абсорбционной фотометрии и их работа?
9. Назначение основных узлов фотометрических приборов?
10. Спектральные характеристики растворов?
11. Принципы выбора аналитических длин волн?
12. Оптимальные условия фотометрических измерений?
13. Фотометрические реагенты и требования, предъявляемые к ним?
14. Приёмы, применяемые для фотометрических измерений?
15. Качественный и количественный анализ в фотометрии?
16. Определение смеси светопоглощающих веществ?
17. Принципы фотометрического титрования?
18. Область фактического применения фотометрических методов анализа?
19. Метрологические характеристики фотометрических методов анализа?
20. Какие из соединений можно определять в УФ-области спектра 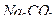 ,
, 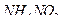 ,
,  ,
,  ?
?
21. Какие из соединений можно определять в видимой области 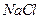 ,
, 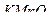 ,
, 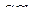 ?
?
22. Предложите оптимальные условия (интервал b и c) для фотометрического определения титана с Н2О2 (молярный коэффициент поглощения равен 720).
23. Вычислите молярный коэффициент поглощения соединения железа, если А = 0,75, b = 5 см, в 50 см3 раствора содержится 0,005 г железа.
24. Рассчитайте оптическую плотность раствора, светопропускание которого равно 60%.
Фототурбидиметрия.
Цель и задачи работы
Изучение работы фотометрических приборов, овладение навыками практического применения фототурбидиметрического и кинетического фототурбидиметрического методов.
Программа работы
Вопросы для самоконтроля
1. В чем заключается суть и отличие методов турбидиметрии и нефелометрии?
2. Приведите примеры применения методов нефелометрии и турбидиметрии в химическом анализе. Перечислите недостатки и достоинства этих металлов.
3. Напишите уравнение Кубелки-Мунка и перечислите следствия, вытекающие из него при измерении спектров, связанных с поглощением света.
4. Перечислите факторы, влияющие на диффузное отражение, погрешности в спектроскопии диффузного отражения. Как измеряют спектры диффузного отражения слабоокрашенных образцов ?
5. Перечислите основные достоинства и недостатки метода оптико-акустической спектроскопии в химическом анализе.
Рефрактометрия
Цель и задачи работы
Изучение работы рефрактометра, овладение навыками практического применения рефрактометрического метода анализа.
Программа работы
Подготовка пробы к анализу:
Мерной пипеткой отбирают в пенициллиновый флакон 5 cм3 свежего молока.
Для осаждения белков к отобранной пробе молока прибавляют 5 капель раствора хлорида кальция, флакон закрывают пробкой и нагревают 10 минут в кипящей водяной бане.
Охлаждают под струёй водопроводной воды до 20о С. Капилляром отбирают несколько капель прозрачной жидкости.
11.2.1.2. Порядок работы на рефрактометре:
1. Перед началом работы прибор проверяют с помощью эталонной плитки с гравированным показателем преломления в проходящем свете. Для этого плитку располагают на измерительной плоскости, увлажненной монобромнафталеном. Среднее значение нескольких определений должно соответствовать значению, выгравированному на плитке.
2. Перед проведением измерений показателя преломления растворов, призмы рефрактометра термостатируют, подключив кожух с помощью двух штуцеров к термостату и пропуская термостатированную воду в течение 15 минут. Контроль за температурой осуществляется с помощью термометра, вставленного в третий штуцер. Температура поддерживается с точностью до 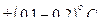 .
.
3. Подняв верхнюю призму, помещают с помощью пипетки 2-3 капли исследуемой жидкости на поверхность горизонтально расположенной нижней призмы. При этом не следует касаться пипеткой поверхности призмы, чтобы не поцарапать её. Затем опускают верхнюю призму и плотно прижимают её к нижней.
4. Перемещая зеркало (осветительную лампу), направляют луч света на систему призм.
5. Резкость устанавливают, вращая окуляр на зрительной трубе.
6. Видимое в зрительной трубе поле делится предельным лучом на светлую и тёмную части, граница между которыми при измерениях должна быть совмещена с перекрестьем нитей или визирной линией вращением барабана.
7. Граница из-за разложения (дисперсии) белого света при прохождении его через измерительную призму может быть нечёткой, размытой и окрашенной во все цвета радуги. Дисперсию устраняют с помощью компенсаторов, например, призмы Амичи, которая состоит из двух призм прямого времени. Призмы вращаются в противоположные стороны и установлены так, что их дисперсия компенсирует дисперсию жидкости и призм рефрактометра.
8. Совместив границы между светом и тенью с центром перекрестья нитей или визирной линией, по шкале отсчитывают показатель преломления
11.2.1.3. Контрольная задача Определение содержания сахара в анализируемом образце:
Быстро (во избежание испарения влаги) наносят 2-3 капли жидкости из прозрачного слоя на нижнюю призму рефрактометра, затем опускают верхнюю призму.
Измерения проводят при 17,5оС, поддерживая эту температуру с помощью термостата.
Выполняют 3-4 параллельных опыта и рассчитывают среднее арифметическое значение показателя преломления.
По таблице 11.1 находят содержание сахара.
Зависимость показателя преломления раствора от массовой доли сахара.
Таблица 11.1.
| Показатель преломления при 17,5оС | Содержание сахара, % | Показатель преломления при 17,5оС | Содержание сахара, % |
| 1,3406 | 3,77 | 1,3420 | 4,49 |
| 1,3408 | 3,87 | 1,3422 | 4,59 |
| 1,3410 | 3,98 | 1,3424 | 4,69 |
| 1,3412 | 4,08 | 1,3426 | 4,79 |
| 1,3414 | 4,18 | 1,3428 | 4,89 |
| 1,3416 | 4,23 | 1,3430 | 4,50 |
| 1,3418 | 4,38 | 1,3432 | 5,10 |
Потенциометрия
Цель и задачи работы
Изучение возможностей и аппаратурного оформления прямой потенциометрии и овладение приемами практического применения потенциометрического титрования.
Программа работы
Подготовка анализируемого образца к титрованию
Проводят по пункту 12.2.1.2.
Подготовка анализируемого образца к титрованию
Анализируемый раствор, содержащий 10-20 см3 раствора сульфата железа (II) и 5-10 см3 хлорида железа (III) переносят в мерную колбу вместимостью 100 см3.
Приливают мерным цилиндром 10 см3 раствора серной кислоты и доводят до метки дистиллированной водой.
12.2.3.2. Подготовка иономера к работе в режиме определения потенциала
1. Платиновый и хлорсеребрянный электроды присоединяют к гнездам "Изм." и "Всп." соответственно и опускают в ячейку с анализируемым растворим.
2. Нажимают на кнопку "t" и вращением ручки "температура раствора" устанавливают по верхней шкале температуру, соответствующую комнатной.
3. Нажимают кнопки "—" и "mV".
4. Выбирают диапазон измерений нажатием кнопок "-1-19" (грубо) или один из точных диапазонов "-1-4"; "4-9"; "9-14"; "14-19".
5. Значения потенциала определяют по нижней (грубо) или верхним (точно) шкалам иономера.
Таблица 12.2
| № точки | Объем раствора титранта V, см3 | Значение потенциала Е, mV | 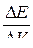 , mV/см3 , mV/см3
|
4. Строят кривые титрования в координатах 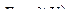 и
и 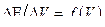 .
.
5. Находят среднее значение объема раствора бихромата калия, израсходованного на титрование сульфата железа (II), и рассчитывают содержание железа (II) в анализируемом растворе в граммах по формуле:
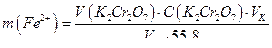
где  - содержание железа (II) в граммах;
- содержание железа (II) в граммах; 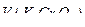 - объем бихромата калия, пошедший на титрование в см3;
- объем бихромата калия, пошедший на титрование в см3; 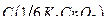 – молярная концентрация эквивалентов бихромата калия, моль/дм3;
– молярная концентрация эквивалентов бихромата калия, моль/дм3;  – объем колбы, см3;
– объем колбы, см3;
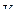 – объем аликвоты, см3.
– объем аликвоты, см3.
Подготовка анализируемого образца к титрованию
Анализируемый раствор хлорида железа (III) переносят в мерную колбу вместимостью 100 см3 и доводят до метки дистиллированной водой.
В стакан для титрования отбирают пипеткой 10 см3 полученного раствора, добавляют 10 см3 раствора соляной кислоты, погружают электроды и доливают дистиллированную воду (электроды должны быть погружены в раствор).
12.2.4.2. Подготовка иономера к работе в режиме определения потенциала.
Проводится по пункту 12.2.3.2.
Подготовка иономера к работе
1. 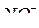 -– селективный электрод предварительно выдерживают в растворе нитрата калия С(1/1KNO3 ) = 0,01 моль/дм3 и хранят в нем.
-– селективный электрод предварительно выдерживают в растворе нитрата калия С(1/1KNO3 ) = 0,01 моль/дм3 и хранят в нем.
2. Измерительный – селективный электрод включают в гнездо «Изм.» на задней стенке прибора с помощью штуцера.
3. Вспомогательный хлорсеребряный электрод включают в гнездо «Всп.».
4. Включают прибор в сеть и нажимают клавиши «t» и «-1 – 19».
5. Прогревают прибор 20-30 мин.
Кулонометрия
Цель и задачи работы
Изучение возможностей и аппаратурного оформления кулонометрии, овладение приемами практического применения кулонометрического титрования в амперостатическом режиме и прямой кулонометрии в потенциостатическом режиме.
Программа работы
Подготовка анализируемого образца HCl к титрованию
Испытуемый раствор переносят в мерную колбу объемом 100 см3 и доводят его объем до метки дистиллированной водой.
Подготовка к работе установки для кулонометрического титрования в амперостатическом режиме
1. В катодное пространство ячейки помещают 25 см3 раствора сульфата калия и доводят объем раствора до 50 см3 дистиллированной водой.
2. Заполняют анодную камеру раствором сульфата калия.
3. Генераторный и стеклянный электроды помещают в катодное пространство, а вспомогательный — в анодную камеру титрационной ячейки. Электроды закрепляют таким образом, чтобы все они были погружены в раствор не менее чем на 1 см.
4. Заполняют соединительный мостик насыщенным раствором хлорида калия и помещают один его конец в катодную камеру ячейки, а другой - в стакан с насыщенным раствором хлорида калия. В этот стакан помещают хлорсеребряный электрод сравнения и закрепляют его таким образом, чтобы он был погружен в раствор не менее чем на 1 см.
5. Стеклянный и хлорсеребряный электроды присоединяют к гнездам "Изм." и "Всп." соответственно иономера.
6. Генераторный и вспомогательный электроды присоединяют к "- " и "+" соответственно источника тока.
7. Для работы источника постоянного тока в режиме стабилизации тока устанавливают кодовый переключатель выходного тока в положение, соответствующее 4 мА, а переключатель напряжения в положение 299 В.
8. Опускают в катодное пространство ячейки якорь магнитной мешалки и включают перемешивание.
Проведение предэлектролиза
1. Перед выполнением контрольной задачи фиксируют значение рН фонового электролита (оно должно быть не более 6). В случае необходимости доводят рН раствора до 6 (вносят 1-3 см3 раствора 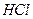 ).
).
2. Включают перемешивание. Тумблер ВКЛ источника постоянного тока устанавливают в верхнее положение и поводят предэлектролиз фонового раствора до тех пор, пока 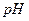 раствора не достигнет
раствора не достигнет 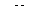 .В этот момент выключают ток тумблером ВКЛ.
.В этот момент выключают ток тумблером ВКЛ.
Подготовка анализируемого образца HCl к титрованию
Проводится по пункту 13.2.1.1.
Подготовка к работе установки для кулонометрического титрования в амперостатическом режиме
1. В катодное пространство ячейки помещают 25 см3 раствора сульфата калия и доводят объем раствора до 50 см3 дистиллированной водой.
2. Заполняют анодную камеру раствором сульфата калия.
3. Генераторный электрод помещают в катодное пространство, а вспомогательный - в анодную камеру титрационной ячейки. Электроды закрепляют таким образом, чтобы они были погружены в раствор не менее чем на 1 см. погружен в раствор не менее чем на 1 см.
4. Генераторный и вспомогательный электроды присоединяют к "- " и "+" соответственно источника тока.
5. Для работы источника постоянного тока в режиме стабилизации тока устанавливают кодовый переключатель выходного тока в положение, соответствующее 4 мА, а переключатель напряжения в положение 299 В.
6. Опускают в катодное пространство ячейки якорь магнитной мешалки и включают перемешивание.
7. В катодное пространство приливают 2-3 капли фенолфталеина.
Проведение предэлектролиза
Тумблер ВКЛ источника постоянного тока устанавливают в верхнее положение и поводят предэлектролиз фонового раствора до тех пор, пока раствор не окрасится в бледно-розовый цвет. В этот момент выключают ток тумблером ВКЛ.
Подготовка пробы к анализу
Испытуемый раствор переносят в мерную колбу объемом 100 см3, доводят его объем до метки дистиллированной водой и тщательно перемешивают.
Проведение предэлектролиза
1. В анодное пространство ячейки наливают 40 смз раствора серной кислоты, погружают рабочий электрод – платиновую сетку и якорь магнитной мешалки. Включают перемешивание.
2. На источнике начального напряжения блока БЗН потенциостата устанавливают выбранный потенциал рабочего электрода (U 1 или U 2) и включают выходное напряжение.
3. Предэлектролиз фона проводят в течение 15 мин.
13.2.4.4. Контрольная задача. Определение содержания 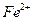 в анализируемом образце
в анализируемом образце
1. Отключают ячейку. Быстро вводят пипеткой в ячейку 0,5 см3 анализируемого раствора.
2. Одновременно включают ячейку и секундомер.
3. Записывают значения миллиамперметра через каждые 10 сек. (таблица 6.5), строят графики зависимости  и
и  .
.
Данные потенциостатической кулонометрии
Таблица 13.5.
| № точки | Время, сек | Показания миллиамперметра, I, mV | 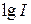
|
4. По графику определяют 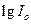 (как отрезок отсекаемый продолжением прямой на оси ординат) и
(как отрезок отсекаемый продолжением прямой на оси ординат) и 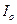 . Для трех произвольных точек рассчитывают
. Для трех произвольных точек рассчитывают 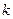 , где
, где 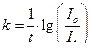 .
.
5. Рассчитывают среднее значение k и количество электричества по формуле:
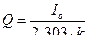
6. Рассчитывают содержание ионов в мг в анализируемом образце по формуле:
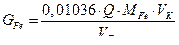
где 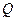 – количество электричества,
– количество электричества,
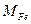 – атомная масса Fe, г/моль;
– атомная масса Fe, г/моль;
 – объем мерной колбы, см 3 ;
– объем мерной колбы, см 3 ;
 - объем аликвоты испытуемого раствора, см3 .
- объем аликвоты испытуемого раствора, см3 .
13.3. Вопросы для самоподготовки
1. В чем различие методов прямой кулонометрии и кулонометрического титрования?
2. Какие законы лежат в основе метода кулонометрии? Сформулируйте их.
3. Каковы особенности метода кулонометрического анализа в амперостатическом и потенциостатическом режимах?
4. Привести принципиальную схему установки для кулонометрического титрования.
5. Назвать наиболее распространенные способы фиксирования точки эквивалентности в кулонометрическом титровании.
6. Привести примеры кулонометрического титрования: а) электрогенерированными окислителями; б) электрогенерированными восстановителями; в) с использованием реакций осаждения и комплексообразования; г) с использованием реакций кислотно-основного взаимодействия.
7. Укажите достоинства и недостатки кулонометрических методов анализа. Чем ограничена область применения этих методов?
Кондуктометрия.
Цель и задачи работы.
Изучение возможностей и аппаратурного оформления кондуктометрии, овладение приемами практического применения кондуктометрического титрования.
Программа работы.
Вопросы для самоконтроля.
1. В чем различие прямой и косвенной кондуктометрии? Какой метод более селективен? Почему?
2. В каком современнном методе анализа используют кондуктометрические детекторы?
3. Электропроводность раствора азотной кислоты, как показали соответствующие измерения, составляет 0,02273 см. Константа ячейки, в которой проводили измерения, была определена в специальном эксперименте. Рассчитайте концентрацию азотной кислоты в растворе, если константа ячейки равна 0,0053 м-1.
4. Растворимость 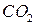 в воде при
в воде при  и давлении 1 атм составляет 1,45 г/дм3. Используя соответствующие величины предельной подвижности ионов и удельной электропроводности воды, находящейся в равновесии с воздухом, рассчитайте степень диссоциации диоксида углерода в воде при . Мольная доля в сухом воздухе равна 0,000314.
и давлении 1 атм составляет 1,45 г/дм3. Используя соответствующие величины предельной подвижности ионов и удельной электропроводности воды, находящейся в равновесии с воздухом, рассчитайте степень диссоциации диоксида углерода в воде при . Мольная доля в сухом воздухе равна 0,000314.
5. Кондуктометрическое титрование 10 см3 0,01 моль/дм3 раствора 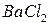 проводилось 0,01 моль/дм3 стандартным водным раствором
проводилось 0,01 моль/дм3 стандартным водным раствором 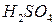 . Рассчитайте удельную электропроводность раствора а) перед началом титрования до добавления серной кислоты, б) в точке эквивалентности и в) после добавления к раствору вдвое большего по сравнению с стехиометрическим количеством . Суммарным ионным эффектом можно пренебречь. Обсудите, насколько целесообразно такое титрование.
. Рассчитайте удельную электропроводность раствора а) перед началом титрования до добавления серной кислоты, б) в точке эквивалентности и в) после добавления к раствору вдвое большего по сравнению с стехиометрическим количеством . Суммарным ионным эффектом можно пренебречь. Обсудите, насколько целесообразно такое титрование.
6. Произведение растворимости сульфата свинца (II) равно приблизительно 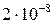 моль2/дм6 . Рассчитайте удельную электропроводность насыщенного раствора сульфата свинца (II), полагая, что при нейтральном рН ионы существуют в растворе в виде простых ионов.
моль2/дм6 . Рассчитайте удельную электропроводность насыщенного раствора сульфата свинца (II), полагая, что при нейтральном рН ионы существуют в растворе в виде простых ионов.
Таблица 15.2.
| № опыта | Объем раствора титранта, V, см3 | Сила тока, I, 10-6 А | 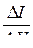
|
11. Строят дифференциальную кривую титрования в координатах 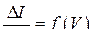 .
.
12. По кривой титрования определяют объем раствора титранта гексацианоферрата (II) калия в точке эквивалетности - 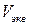 .
.
13. Содержание цинка рассчитывают по формуле:
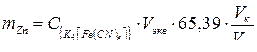 ,
,
где 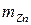 -. масса цинка, г;
-. масса цинка, г; 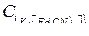 - молярное содержание раствора гексацианоферрата (II) калия, моль/дм3; - объем раствора гексацианоферрата (II) калия в точке эквивалетности;
- молярное содержание раствора гексацианоферрата (II) калия, моль/дм3; - объем раствора гексацианоферрата (II) калия в точке эквивалетности;  - объем колбы;
- объем колбы; 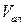 - объем аликвотной части раствора соли цинка, взятый для титрования.
- объем аликвотной части раствора соли цинка, взятый для титрования.
Вопросы для самоконтроля.
1. Как выбирают условия проведения амперометрического титрования ? Почему графитовый или платиновый электрод в амперометрическом титровании используют чаще, чем ртутный капающий ?
2. От чего зависит вид кривой амперометрического титрования ?
3. В кислой сульфатной среде реакция между ванадием (IV) и ванадием (II) с образованием ванадия (III) протекает количественно, поэтому её можно использовать для амперометрического титрования ванадия (IV). Ванадий (II) даёт анодную волну с потенциалом полуволны –51 В (относительно нас. КЭ), а V (IV) даёт катодную волну с потенциалом полуволны –0,85 В. Выберите подходящий потенциал для амперометрического титрования 10 см3 1,0 ммоль/дм3 раствора ванадия (IV) 2,0 ммоль/дм3 раствором ванадия (II) и покажите, какой должна быть кривая титрования, полученная при выбранном потенциале.
Хроматографический анализ
Цель и задачи работы
Изучение принципов основных методов хроматографического анализа, различающихся механизмом реакции, способом элюирования и аппаратурным оформлением. Овладение навыками осуществления обнаружения, идентификации и определения компонентов пробы (решение практических задач).
Программа работы
Результаты определений
Таблица 16.3.
| № стандарта | Концентрация иона, г-экв/дм3 | Размер зоны | |
| мм | см3 | ||
Подготовка капилляра
Капиллярную трубку диаметром 0,5 мм оттягивают с одного конца на газовой горелке в виде конуса и обрезают капилляр длиной 25-30 мм. Нижний торец капилляра отшлифовывают. Затем капилляр тщательно промывают эфиром, этиловым спиртом и дистиллированной водой. Чистый капилляр заполняется водой мгновенно. Если этого не происходит, то промывание повторяют. Хранят капилляр в дистиллированной воде.
Для калибровки капилляра его наполняют дистиллированной водой, наружные стенки вытирают фильтровальной бумагой и взвешивают на аналитических весах. Затем удаляют воду из капилляра прикосновением фильтровальной бумаги и снова взвешивают. По разности результатов взвешиваний определяют емкость капилляра.
Капельный анализ неорганических веществ
Цель и задачи работы
Овладеть приемами выполнения качественных аналитических реакций капельным способом. Освоить методику дробного качественного анализа, обнаружения катионов и анионов в многокомпонентных средах с помощью специфических реакций. Произвести анализ объекта неизвестного состава.
Программа работы
1.2.1. Предварительная подготовка пробы
Анализ твердых объектов
Пробу измельчают в мельнице или растиранием в ступке. Берут навеску 0,1-0,5 г и растворяют в соответствующем, в зависимости от предполагаемого химического состава, растворителе (вода, растворы кислот и щелочей).
1.2.1.2. Анализ металлов и сплавов (безстружковый метод)
На очищенную поверхность образца помещают 2-3 капли азотной кислоты (1:1). После прекращения реакции, образовавшийся на поверхности металла раствор разбавляют дистиллированной водой (2-3 капли) и переносят с помощью капилляра или пипетки в микропробирку. Операцию повторяют 2-3 раза. Собранный раствор подвергают непосредственно анализу.
Анализ жидких сред
В зависимости от компонентного состава пробы, при необходимости, по специальным методикам проводят операции маскирования, осаждения и отделения органических соединений и отдельных ионов, мешающих анализу, или выделения открываемых ионов.
Дата: 2018-12-28, просмотров: 809.