БАШКИРСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
ГИЗАТУЛЛИН А.Г.
КРАТКАЯ ПАТОФИЗИОЛОГИЯ
ОРГАНОВ И СИСТЕМ
ЛЕКЦИИ
(Частная патофизиология)
Г .
ОГЛАВЛЕНИЕ
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ ДЕЯТЕЛЬНОСТИ.. 2
АРТЕРИАЛЬНЫЕ ГИПЕРТЕНЗИИ.. 7
АРТЕРИАЛЬНЫЕ ГИПОТЕНЗИИ.. 10
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ ДЫХАНИЯ.. 12
ПАТОФИЗИОЛОГИЯ КРОВИ.. 17
РЕГУЛЯЦИЯ ЭРИТРОПОЭЗА И ЕГО НАРУШЕНИЯ.. 19
АНЕМИИ.. 19
ЛЕЙКОЦИТОЗЫ И ЛЕЙКОПЕНИИ.. 22
ЛЕЙКОЗЫ... 24
ПАТОФИЗИОЛОГИЯ ГЕМОСТАЗА.. 27
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ ПИЩЕВАРЕНИЯ.. 30
ПАТОФИЗИОЛОГИЯ ПЕЧЕНИ.. 34
ПАТОФИЗИОЛОГИЯ ПОЧЕК.. 39
ПАТОФИЗИОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ... 44
БОЛЕЗНИ ГИПОТАЛАМО-ГИПОФИЗАРНОЙ СИСТЕМЫ... 44
АКРОМЕГАЛИЯ.. 44
НЕДОСТАТОЧНОСТЬ АДЕНОГИПОФИЗА.. 44
ГИПОФИЗАРНЫЙ НАНИЗМ... 45
СИНДРОМ И БОЛЕЗНЬ ИЦЕНКО—КУШИНГА.. 45
НЕСАХАРНЫЙ ДИАБЕТ.. 46
ЗАБОЛЕВАНИЯ НАДПОЧЕЧНИКОВ.. 46
ПЕРВИЧНЫЙ ГИПЕРАЛЬДОСТЕРОНИЗМ... 46
ВТОРИЧНАЯ НЕДОСТАТОЧНОСТЬ КОРЫ НАДПОЧЕЧНИКОВ.. 46
ФЕОХРОМОЦИТОМА.. 46
ЗАБОЛЕВАНИЯ ЩИТОВИДНОЙ И ПАРОЩИТОВИДНЫХ ЖЕЛЕЗ. 47
ДИФФУЗНЫЙ ТОКСИЧЕСКИЙ ЗОБ.. 47
ГИПОТИРЕОЗ. 47
ЗАБОЛЕВАНИЯ ПАРОЩИТОВИДНОЙ ЖЕЛЕЗЫ... 48
ПЕРВИЧНЫЙ ГИПЕРПАРАТИРЕОЗ. 48
ГИПОПАРАТИРЕОЗ. 48
ЗАБОЛЕВАНИЯ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ... 49
ПЕРВИЧНЫЙ САХАРНЫЙ ДИАБЕТ.. 49
ОСТРЫЕ ОСЛОЖНЕНИЯ ДИАБЕТА.. 50
ЗАБОЛЕВАНИЯ ЖЕНСКИХ ПОЛОВЫХ ЖЕЛЕЗ. 50
ЗАБОЛЕВАНИЯ МУЖСКИХ ПОЛОВЫХ ЖЕЛЕЗ. 51
ПАТОЛОГИЧЕСКАЯ ФИЗИОЛОГИЯ НЕРВНОЙ СИСТЕМЫ... 51
ПАТОФИЗИОЛОГИЯ НЕВРОЗОВ.. 54
ЛИТЕРАТУРА.. 58
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ ДЕЯТЕЛЬНОСТИ
Важнейшим типовым понятием является недостаточность кровообращения - неспособность системы кровообращения обеспечить потребность органов и тканей O2 и субстратами метаболизма.
Понятие патофизиологии кровообращения включает в себя понятия сердечной и сосудистой недостаточности.
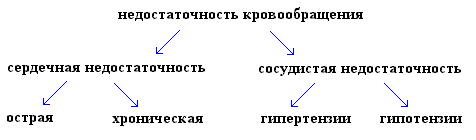
I . Сердечная недостаточность (СН) (insufficientia cordis) – патологическое состояние, обусловленное неспособностью сердца обеспечить адекватное кровоснабжение органов и тканей кровью, т.е. неспособностью перекачать всю поступающую венозную кровь (в отличии от сосудистой недостаточности - недостаток притока к сердцу венозной крови).
Формы и механизмы аритмии:
Аритмии - (отсутствие ритма, неритмичность) - различные изменения основных электрофизиологических характеристик миокарда (автоматизма), ведущие к нарушению нормальной координации сокращений различных участков миокарда или отделов сердца с резким учащением или урежением сердечных сокращений.
IV . Нарушение сократимости миокарда.
V . Нарушение ферментативного спектра миокарда.
Острая сердечная недостаточность (ОСН) - виды, причины и патогенез
Пять форм ОСН:
а) тампонада сердца,
б) полная атрио-вентрикулярная блокада,
в) мерцание, фибрилляция желудочков,
г) инфаркт миокарда и
д) острая закупорка легочной артерии.
Тампонада сердца - синдром острой сердечной недостаточности, вызванной внутриперикардиальным сдавлением сердца жидкостью (гемотампонада, перикардит острый экссудативный) или газом.
Патогенез нарушений:
1) механическое сдавление тонкостенных отделов сердца и крупных вен → уменьшение наполнения его полостей. Развивается синдром низкого сердечного выброса (резкое уменьшение сердечного и МОС), уменьшение тканевого кровотока, олигоурия, повышение потребления, повышение потребления O2 и увеличение содержания в крови молочной и пировиноградной кислоты и
2) патологический вагусный рефлекс - из-за растяжения перикарда → раздражение nervus vagus → может быть остановка сердца, АД понижается, а венозное повышается. При наличии большого выпота, когда резко ограничивается диастола и сильное затруднена работа сердца - возниает кислородное голодание мозга: беспокойство, чувство тревоги. Нарастает бледность кожи и затем цианоз. Это легко воспроизводится в эксперименте.
Полная атрио-вентрикулярная блокада - различают 4 степени атрио-вентрикулярной блокады:
1 степень - удлинение времени предсердно-желудочкового проведения;
2 степень - выпадение некоторых желудочковых комплексов после постепенного удлинения интервала P-Q, но после выпадения желудочкового сокращения проводимость на непродолжительное время улучшается, а затем вновь периоды Венкебаха - Самойлова.
При блокаде 3 степени из предсердий в желудочки проводится лишь каждый 2, 3, 4-й импульс и
4 стадия - полная поперечная блокада.
Причины: гипоксия, тяжелая патология миокарда с нарушением метаболизма, инфаркт миокарда, интоксикация, рубцы, ревматизм. Нет синхронизации сокращений предсердий (60-70/мин) и желудочков (40-50). Если обе систолы вместе - холостое сокращение желудочков и снижение МО. Нарушение предсердно-желудочковой проводимости - частая причина нарушения сердечного ритма, уступающая по частоте лишь экстрасистолии, мерцательной аритмии и пароксизмальной желудочковой тахикардии.
Мерцание желудочков - форма мерцательной аритмии - нарушение ритма сердца с частыми и нерегулярными возбуждениям миокарда и полной разнородностью сердечных сокращений по частоте, силе, причем длительность сердечных циклов значительно колеблется и носит случайный характер. При мерцании частота волн на ЭКГ более 6300/ мин (обычно 500-800/ мин), а при трепетании - менее 300/мин. Глянцевая поверхность миокарда при этом мерцает, напоминая рябь на поверхности воды, в связи с чем это состояние получило название "мерцание" предсердий.
Фибрилляция - наличие сокращений миокардиальных волокон при отсутсвии сокращения всего миокарда как целого. Сердечные волокна сокращаются разрозненно и разновременно не выполняя насосную функцию: УО и МО = 0, гибель через 5 мин, человек не может жить.
Причины: тяжелая гипоксия, ишемия миокарда, интоксикация, нарушение электролитного баланса, повреждение механическое и электротоком, низкая темпиратура, нервно-психическое возбуждение, применение симпатомемитических средств при наркозе.
АРТЕРИАЛЬНЫЕ ГИПЕРТЕНЗИИ
Артериальная гипертензия (АГ) (от греч. hyper – чрезмерный, лат. tensio – напряжение) - стойкое повышение артериального давления - важный симптом патологических состояний и заболеваний, сопровождающихся либо увеличением сопротивления артериальному кровотоку, либо повышением сердечного выброса, либо сочетанием этих факторов. В норме АД=110-140 / 65-90 мм рт. ст., а 150 / 94 - переходная зона, еще не гипертензия.
СДЦ играет главную роль в нейрогуморальной регуляции кровообращения, в нем можно выделить 3 взаимосвязанных отдела:
а) группа нейронов, расположенных в латеральных частях продолговатого мозга - их постоянная активность через пре - и постганглионарные симпатические нейроны оказывает тоническое активизирующее влияние на функцию сердца и гладкой мускулатуры сосудов;
б) медиально расположенные нейроны, обладающие противоположным (тормозным) действием на пре - и постганглионарные симпатические нейроны и уменьшающие влияние адренергической иннервации на кровообращение;
в) дорсально расположенное ядро блуждающего нерва, оказывающего тормозящее влияние на сердце.
Эфферентные механизмы (периферическое звено функциональной системы) реализуются через симпатический отдел нервной системы и эндокринную (гипофиз, надпочечники, щитовидная железа – повышение АД). Но есть и механизмы обратной связи - депрессорный механизм - при растяжении дуги аорты и синокаротидной зоны (при растяжении стенки общей сонной артерии) усиливается депрессорное влияние на СДЦ и тормозит его Длительная или значительная артериальная гипертензия сама по себе формирует патологическое состояние, которое проявляется перегрузкой и гипертрофией сердца, напряжением адаптационных механизмов регионарного кровообращения. Гипертензия - системное повышение давления в артериях большого круга кровообращения, а гипертония - повышение мышечного тонуса - спазмирование сосудов.
Актуальность: высокая частота, ведущее место - гипертоническая болезнь - у 5-6% населения, высокая опасность атеросклероза, инсульта, тромброза сосудов и т.д. Артериальная гипертензия - одна из форм сосудистой недостаточности, как и артериальная гипотензия - сосудистая недостаточность в форме гипотензии (есть еще и сердечная недостаточность - но чаще смешанные формы - сердечно-сосудистая недостаточность).
АРТЕРИАЛЬНЫЕ ГИПОТЕНЗИИ
Гипотензии - понижение артериального давления ниже 100/60 мм рт.ст. (для лиц старше 30 лет - ниже 105/65 мм рт. ст.) или при снижении среднединамического АД ниже 75 (90/60:2=75).
Классификация:
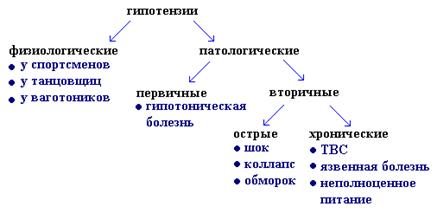
Первичная (эссенциальная) артериальная гипотензия некоторыми авторами рассматривается как гипотоническая болезнь - гипотензивный тип нейро-циркуляторной дистонии. Предполагается, что в патогенезе первичной артериальной гипотонии играют роль нарушения высших вегетативных центров вазомоторной регуляции, это ведет к стойкому уменьшению общего периферического сопротивления при недостаточном увеличении сердечного выброса.
Течение и клиническое проявления первичной артериальной гипотензии не закономерны. Весной и летом чаще обострения, а так же после острых инфекционных заболеваний.
Встречается заболевание чаще у женщин 31-40 лет. При развитии у детей и подростков придают значение наследственной отягощенности, несоблюдению режима дня, конфликтным ситуациям в школе или дома, перенесенным инфекционным заболеваниям.
Острая артериальная гипотензия чаще всего является следствием острой сердечно-сосудистой недостаточности или сосудистой недостаточности при шоке различного генеза или коллапсе, при внутреннем кровотечении и кровопотере, при внезапном перераспределении массы циркулирующей крови, при тяжелых интоксикациях, инфекциях (грипп, сыпной и брюшной тиф, пищевые токсикоинфекции, дизентерия), отравлении различными ядами, при неправильном применении нейролептиков, ганглиоблокаторов, симпатолитиков.
Из островозникающих гипотензий наиболее важное место занимает шок - типовой фазово-развивающийся патологический процесс, возникающий вследствие расстройств нейрогуморальной регуляции, вызванных экстремальными воздействиями и характеризуется уменьшением кровоснабжения тканей, нарушением обменных процессов, гипоксией и угнетением функций организма.
Коллапс - остро развивающаяся сосудистая недостаточность с падением сосудистого тонуса и острым понижением ОЦК. При этом уменьшается приток венозной крови к сердцу, снижается серд. выброс, понижается АД и венозное давление, нарушается перфузия тканей и обмен веществ, развивается гипоксия головного мозга и угнетаются жизненноважные функции организма. Коллапс развивается как осложнение при тяжелых патологических состояниях и острых заболеваниях внутренних органов - при перитоните, остром панкреатите, что связано с эндогенной интоксикацией. Инфекционный коллапс развивается как осложение при острых менингоэнцефалитах, брюшном и сыпном тифе, острой дизентерии, и т.п. в связи с интоксикацией эндо- и экзотоксинами микроорганизмов, преимущественно влияющей на ЦНС или на рецепторы пре- и посткапилляров.
Гипоксический коллапс может быть в условиях понижения O2 особенно в сочетании с пониженным барометрическим давлением. Причиной циркуляторных нарушений здесь будет недостаточность приспособительных реакций организма, что ведет к гипоксии, воздействующей прямо или косвенно через рецепторный аппарат системы на СДЦ. Развитию коллапса способствует гипокапния (следствие гипервентиляции, что ведет к расширению сосудов, депонированию и понижению ОЦК).
Ортостатический коллапс (возникает при быстром переходе из горизонтального в вертикальное положение и обусловлен перераспределением крови с увеличением общего объема венозного русла и снижением венозного возврата к сердцу. В основе его лежит недостаточность венозного тонуса. Он может возникать в послеоперационном периоде при быстрой эвакуации асцитической жидкости или в результате спинномозговой или перидуральной анестезии.
Одной из частых форм является геморрагичесий коллапс, развивающийся при острой массивной кровопотере в связи с быстрым уменьшением ОЦК. Такое же состояние может возникнуть в следствие обильной плазмопотери при ожогах, водно-электролитных расстройствах при тяжелой диарее, неукротимых рвотах, при нерациональном применении мочегонных средств.
Коллапс может быть при острых заболеваниях сердца, сопровождающихся резким и быстрым понижением УО (инфаркт миокарда, гемоперикард, острый миокардит, при тромбоэмболиях легочной артерии). Острая сердечнососудистая недостаточность при этих состояниях часто называется синдром малого выброса (при кардиогенном шоке).
В патогенезе коллапса можно условно выделить два основных механизма, которые часто сочетаются. Один механизм заключается в падении тонуса артериол и вен в результате воздействия инфекционных, токсических, физических, аллергических, и других факторов непосредственно на сосудистую стенку, СДЦ и сосудистые зоны. При недостаточности компенсаторных механизмов снижение периферического тонуса сосудов (парез сосудов) ведет к патологическому повышению емкости сосудистого русла, понижению ОЦК с депонированием крови в некоторых сосудистых областях, понижением венозного притока к сердцу, повышением ЧСС, понижением АД.
Другой механизм связан непосредственно с быстрым понижением массы циркулирующей крови (например, при массивной кровопотере и плазмопотере, когда это превосходит компенсаторные возможности организма). Возникающий в ответ на это рефлекторный спазм мелких сосудов + повышение ЧСС под влиянием повышенного выброса катехоламинов может оказаться недостаточными для сохранения нормального уровня АД. Понижение ОЦК снижает возврат крови к сердцу и соответственно понижает УО, нарушает систему микроциркуляции. Кровь скапливается в капиллярах, понижается АД. Развивается гипоксия циркуляторного типа и метаболический ацидоз, которые ведут к повреждению сосудистой стенки, повышению ее проницаемости. Это способствует переходу воды и электролитов из крови в межклеточное пространство. Нарушаются реологические свойства, возникает гиперкоагуляция крови, патологическая агрегация эритроцитов и тромбоцитов, создаются условия для образования микротромбозов.
Обморок - внезапно развивающееся патологическое состояние характеризуется резким ухудшением самочувствия, тягостными переживаниями, дискомфортом, вегетативно-сосудистыми расстройствами, снижением мышечного тонуса и кратковременным нарушением сознания. Обморок может быть вызван действием различных факторов, вызывающих преходящий спазм сосудов головного мозга, в том числе отрицательными эмоциями в связи с испугом, неприятным зрелищем, в конфликтной ситуации (психогенный обморок), болью (болевой обморок). Клинически обморок имеет три последовательные сменяющиеся стадии - предвестников, нарушение сознания и восстановительный период.
Лечение направлено на улучшение кровоснабжения и оксигенации головного мозга. Больного нужно положить, опустить голову и приподнять ноги, обеспечить доступ свежего воздуха, освободить от стесняющей одежды, побрызгать в лицо холодной водой. При необходимости дать подышать парами нашатырного спирта.
В тяжелых случаях, когда обморок затягивается - показан непрямой массаж сердца и искуственое дыхание.
Причины и механизмы одышки:
1. Гиперкапния - повышение содержания CO2 в артериальной крови. В норме парциальное давление (pCO2) составляет 38-40 мм рт. ст. и является очень постоянной величиной, как и pH крови. Повышение pCO2 артериальной крови всего на 2 мм рт. ст. ведет к увеличению легочной вентиляции на 10 л/мин, и нормализации pCO2.
2. Снижение pO2 в крови ведет к гипоксии и возбуждению дыхательного центра - гипервентиляции и вымыванию CO2 из крови. При этом чувствительность дыхательного центра к CO2 резко повышается. В результате при возвращении к условиям нормального атмосферного давления возникает стадия одышки. Такое явление может быть после искусственной гипервентиляции на ИВЛ при реанимации. Еще более важным этиологическим фактором в механизме одышки является гипоксемия, вызывающая резкое возбуждение дыхательного центра. Одновременно возникает нарушение функции коры головного мозга. В результате поступающая в кору головного мозга импульсация из дыхательного центра оценивается неадекватно, извращенно.
Виды одышки:
1) полипноэ - частое и глубокое дыхание при болевом раздражении, мышечной работе. Имеет компенсаторное значение.
2) тахипное - частое, но поверхностное дыхание при раздражении альвеол легких, при пневмонии, отеке и застойных явлениях.
3) брадипноэ - глубокое и редкое дыхание (стенотическое) при затруднении прохождения воздуха через верхние дыхательные пути, трахею, бронхи. Альвеолы заполняются медленно, раздражение их слабое и медленно наступает смена вдоха на выдох (замедление рефлекса Геринга-Брейера).
4) апноэ - остановка дыхания.
Если при одышке затруднен вдох - инспираторная - при затруднении прохождения воздуха через ВДП (истинный круп при дифтерии, закупорка бронха).
При затруднении выдоха - экспираторная - характерна при поражении легочной ткани, особенно при потере ее эластичности (эмфизема легких).
Нередко одышка бывает смешанная - когда затруднен вдох и выдох.
Отек легких (oedema pulmonum) - тяжелое патологическое состояние, обусловленное обильным пропотеванием жидкой части крови в интерстициальную ткань легких, а затем и в альвеолы, ведущее к тяжелому удушью, цианозу, клокочущему дыханию, асфиксии и гибели организма.
Смертность при отеке легких составляет от 20 до 50%. А при развитии отека легких в остром периоде инфаркта миокарда, осложненного кардиогенным шоком или при сочетании отека легких с анафилактическим шоком смертность достигает 90 %.
По скорости развития отека легких различают:
Молниеносную форму, которая заканчивается гибелью организма в течение нескольких минут.
Острый отек легких, продолжающийся 2 - 4 часа и затяжной отек легких, может длиться несколько суток.
Этиология отека легких:
1) Недостаточность левого желудочка сердца, ведущая к резкому подъему давления в легочных капиллярах в результате застоя крови в малом круге кровообращения – так называемые кардиогенные факторы. На первом месте стоит инфаркт миокарда. Пропотевание богатой белком жидкости в легочную ткань возникает тогда, когда гидростатическое давление в капиллярах легких достигает уровня коллоидно-осмотического давления крови и особенно, если превышает его. Если же этому предшествует гипоксия капиллярной стенки, то наступает повышение проницаемости капилляров и отек легких развивается при более низком гидростатическом давлении.
Клинически отеку легких предшествуют приступы сердечной астмы (особенно ночью во время сна) в результате усиления деятельности правого желудочка и повышения кровенаполнения легких венозной кровью.
2) Введение большого количества (до нескольких литров) крове- и плазмозаменителей (после кровопотери) без соответствующего контроля за диурезом. Особенно опасно излишнее введение безбелковых жидкостей - физраствора, который не только повышает гидростатическое давление, но и понижает коллоидно-осмотическое давление крови.
3) Резкое снижение давления в плевральной полости (после быстрого удаления плеврального транссудата).
4) Различные интоксикации, вызывающие повышение проницаемости сосудистой и альвеолярной стенки: ● при острых пневмониях; вдыхании токсических агентов (например, боевых отравляющих веществ удушающего действия - фосген, дифосген, угарный газ);
● диффузное поражение капиллярной стенки эндотоксинами при тяжелых инфекционных заболеваниях (тифы, грипп, дифтерия).
● почечный отек легкого при недостаточности функции почек, печеночный, действие вазоактивных соединений - гистамин, серотонин, простагландин.
5) Альвеолярная гипоксия, вызывающая нарушение тонуса легочных сосудов, который регулируется парциальным давлением кислорода в альвеолах. Это может быть при глубоком угнетении дыхательного центра (наркоз, отравление снотворными, психотропными веществами и т.п., при кровоизлияниях в мозг, опухолях, менингитах, энцефалитах, эклампсии).
6) Аллергический отек легких возникает по типу аллергии немедленного типа, обычно сопутствует анафилактическому шоку, развивается молниеносно и приводит к гибели организма в течение нескольких минут.
Патогенез отека легких. В патогенезе отека легких основное значение имеют следующие факторы:
● острое повышение гидростатического давления в капиллярах малого круга кровообращения;
● повышение проницаемости капиллярной стенки;
● снижение коллоидно-осмотического давления плазмы крови;
● быстрое падение внутриплеврального давления;
● нарушение центральной и рефлекторной регуляции:
а) тонуса сосудов легких,
б) проницаемости сосудистой стенки, скорости кровотока, лимфооттока от легочной ткани.
Все эти факторы обычно действуют в сочетании друг с другом. В то же время каждый из них может иметь ведущее значение.
В динамике отека легких выделяют 2 фазы:
I фаза - интрамуральная (или интерстициальная) характеризуется очаговым спазмом или наоборот паралитическим расширением капилляров, набуханием альвеол или дыхательного эпителия пневмоцитов I или II порядка, пропитыванием межальвеолярных перегородок отечной жидкостью с образованием пузырей и отслоением эпителия альвеол. Все это ведет к увеличению толщины межальвеолярных перегородок в 3-4 раза.
2 фаза - альвеолярного отека характеризуется накоплением жидкости уже в просвете альвеол. При этом происходит дальнейшее поражение альвеолярной стенки: вакуолизация и расплавление волокон, дистрофия эндотелия капилляров и пневмоцитов, их слущивание и разрушение.
Клинически отек легкого проявляется сильнейшей одышкой. Частота дыхания достигает 30-40/мин. Быстро появляется акроцианоз. Дыхание становится клокочущим, слышно на расстоянии. Выделяется обильная пенистая мокрота, наступает возбуждение, страх смерти. Человек тонет в собственной жидкости. Оказание экстренной помощи при отеке легких включает различные направления:
1. Борьба с пенообразованием:
а) дыхание кислородом, увлажненным спиртом и
б) применение специальных пеногасителей.
2. Для разгрузки сердечной деятельности необходимо уменьшение объема циркулирующей крови:
а) наложение жгутов на конечности,
б) дозированное кровопускание и
в) применение мочегонных средств.
Виды повреждения плевры.
Наиболее частым повреждением плевральной полости являются:
● пневмоторакс - попадание воздуха в плевральную полость;
● гидроторакс - скопление транссудата или экссудата;
● гемоторакс - кровоизлияние в плевральную полость.
Особенно опасен пневмоторакс, требующий срочного оказания помощи, иначе пострадавший может погибнуть.
Виды пневмоторакса:
1. естественный при попадании воздуха в плевральную полость при разрушении бронхов и бронхиол например при туберкулезе, асбцессах, гангрене, раке, актиномикозе и особенно при гнойном воспалении с образованием свища.
2. искусственный:
а) при ранениях и повреждении грудной клетки;
б) лечебный при инфильтративном или кавернозном туберкулезе с целью создания покоя и мобилизации РЭС.
Пневмоторакс может быть односторонним и двусторонним.
По степени заполнения плевральной полости и сдавления (точнее спадения легкого) различают: частичный (часть легкого спадается) и полный (полное спадение) легкого. Полный двусторонний пневмоторакс не совместим с жизнью.
По характеру сообщения с окружающей средой различают:
а) закрытый пневмоторакс - когда после попадания воздуха в плевральную полость отверстие сразу же закрывается и сообщение с атмосферным воздухом прекращается. Такой пневмоторакс переносится сравнительно легко, т.к. воздух всасывается, спадение легкого уменьшается и оно вновь участвует в дыхании.
б) открытый пневмоторакс - когда воздух свободно входит и выходит при каждом вдохе и выдохе. При этом давление в плевральной полости равно атмосферному, легкое поджимается к корню и выключается из дыхания. А т.к.с каждым вдохом входит новая порция воздуха, легкие не расправляются до тех пор, пока открытый пневмоторакс не будет переведен в закрытый и не наступит рассасывание воздуха. Поэтому при оказании помощи при ранении грудной клетки необходимо любым путем срочно перевести открытый пневмоторакс в закрытый.
в) клапанный или напряженный пневмоторакс возникает, когда на месте отверстия из тканей мышц или плевры образуется обрывок ткани, двигающийся подобно клапану. И во время вдоха воздух присасывается в плевральную полость, а во время выдоха отверстие закрывается клапаном и воздух обратно не выходит. Клапанный пневмоторакс протекает наиболее тяжело и даже односторонний пневмоторакс может привести к гибели не только из-за полного спадения легкого, но и возникающего смещения средостения - сдавление сердца, сосудов и другого легкого.
Оказание экстренной помощи при клапанном пневмотораксе заключается в удалении воздуха из плевральной полости с последующей герметизацией ее. Для этого клапанный пневмоторакс сначала переводят в открытый, вводя иглу в плевральную полость во втором межреберье и одновременно накладывают окклюзионную повязку на рану грудной стенки. После удаления воздуха органы средостения возвращаются на свои места, после чего иглу сразу же выводят из полости плевры.
ПАТОФИЗИОЛОГИЯ КРОВИ
В норме у человека 4-5 л. С учетом изменения гематокрита - соотношения объема форменных элементов крови (у мужчин 45%, у женщин 42%) к объему плазмы выделяют 3 варианта изменений объема крови (ОК):
1. гиповолемия - при потере крови, сгущение крови;
2. гиперволемия - введение жидкости, увеличение производства эритроцитов;
3. нормоволемия - объем крови не изменен, но изменен гематокрит
По содержанию форменных элементов крови выделяют:
а) олигоцитемичекие,
б) нормоцитемические,
в) полицитемические.
АНЕМИИ
Анемии это состояния, характеризующиеся уменьшением количества эритроцитов, или гемоглобина, или эритроцитов и гемоглобина в единице объема крови.
Классификация анемий:
1. При сравнительной оценке снижения количества эритроцитов и количества Hb основным тестом является цветовой показатель (ЦП) - среднее содержание Hb в одном эритроците. По этому признаку различают следующие виды анемий:
а) Нормохромная - с нормальным (0.9-1.0) цветовым показателем. Этот вариант анемии свидетельствует о пропорциональном, равномерном снижении Hb и эритроцитов в единице объема крови.
б) Гипохромная - со снижением (менее 0.9) ЦП. Этот вид анемии свидетельствует о том, что количество Hb снижено в большей степени, чем эритроцитов
в) Гиперхромная - с повышением (более 1.0) ЦП. Этот вид анемии встречается в тех случаях, когда общее количество эритроцитов снижено в большей степени, чем общее количество Hb или в крови присутствуют клетки эмбрионального типа кровотворения (мегалоциты).
2. По регенераторной способности костного мозга:
а) регенераторные;
б) гиперрегенераторные;
в) гипорегенераторные;
г) арегенераторные.
3. По размеру эритроцитов:
а) нормоцитарные;
б) макроцитарные;
в) микроцитарные.
4. По типу кроветворения:
а) нормобластическая - с эритробластическим типом кроветворения,
б) мегалобластическая - с мегалобластическим типом кроветворения,
5. По этиологии:
а) постгеморрагические - вследствии кровопотерь,
б) гемолитические - вследствии повышенного кроворазрушения,
в)вследствии нарушения кровообразования - B12-дефицитная и фолиеводефицитная анемии, железодефицитная анемия и др.
Постгеморрагические анемии могут быть острыми и хроническими.
Острая постгеморрагическая анемия возникает после одномоментной, быстрой массивной кровопотери. Такая ситуация возникает при ранении крупных сосудов, кровотечениях из внутренних органов и выделяют следующие стадии:
В первые часы после острой кровопотери наблюдается относительно равномерное уменьшение количества эритроцитов и Hb, цветовой показатель (ЦП) в пределах нормы (нормохромная анемия).
На 2-3 сутки количество эритроцитов уменьшается за счет поступления тканевой жидкости в сосуды ( относительная эритропения) и разрушения эритроцитов в клетках системы мононуклеарных фагоцитов (абсолютная эритропения).
На 4-5 день усиливается эритропоэз за счет выработки эритропоэтина. В периферической крови увеличивается количество полихроматофильных эритроцитов (ретикулоцитов), появляются нормобласты (регенераторня анемия), ЦП снижается (гипохромная анемия), т.к. ускореннная регенерация опережает созревание клеток, которые не успевают потерять признаки своей незрелости (ядро, гранулы) и насытиться Hb. Острая кровопотеря приведет к дефициту железа и снижению синтеза гема.
Хронические постгеморрагические анемии возникают при небольших по объему, но частых и длительных кровотечениях (при язвенной болезни желудка, геморрое, гиперполименорее и т.д.), при нарушении гемостаза (геморрагический диатез). К этой категории анемии относится также анкилостомная анемия, развивающаяся при инвазии паразитами из класса нематод. Паразит прикрепляется к стенке тонкой кишки и питается кровью хозяина. Это вызывает травматизацию кишечной стенки и кровотечение.
Основным гематологическим признаком хронической постгемаррогической анемии является выраженная гипохромия эритроцитов, которая свидетельствует о резком снижениии синтеза Hb из-за дефицита железа. Хронические потери крови приводят к истощению депо железа, поэтому хронические постгемаррогические анемии всегда железодефицитные. Для такой анемии характерен микроцитоз. При угнетени кроветворения эта анемия может быть гипо - и арегенераторной.
Гемолитические анемии - характеризуются преобладанием процессов разрушения эритроцитов над процессом их образования. Усиление распада эритроцитов может быть обусловлено:
приобретенными или наследственными изменениями метаболизма и структуры мембраны, стромы эритроцитов или молекул Hb;
повреждающим действием физических, химическхе, биологических гемолитических факторов на мембрану эритроцитов;
замедлением движения эритроцитов в межсинусовых пространствах селезенки, что способствует их разрушению макрофагоцитами;
усилением активности макрофагоцитов.
● Наследственные гемолитические анемии:
1. Эритроцитопатии:
a) нарушение структуры оболочки с изменением формы (наследственный микросфероцитоз или анемия.Минковского-Шоффара, наследственный овалоцитоз);
б) энзимопатии – дефицит ферментов пентозофосфатного цикла, гликолиза и др. (глюкозо-6-фосфат-дегидрогеназы).
2. Гемоглобинопатии:
а) наследственный дефект синтеза цепей глобина (d- и b-талассемия);
б) наследственный дефект первичной структуры глобина (серповидноклеточная анемия).
● Приобретенные гемолитические анемии:
а) токсическая (гемолитические яды: соединения мышьяка, свинца; токсины возбудителей инфекций: гемолитический стрептококк, анаэробный малярийный плазмодий);
б) имунная (переливание несовместимой крови, Rh-несовместимость матери и плода; образование аутоантител против собственных эритроцитов при изменении их антигенных свойств под влиянием лекарств, вирусов);
в) механическая (механическое повреждение эритроцитов при протезировании сосудов, клапанов);
г) приобретенная мембранопатия (соматическая мутация под действием вирусов, лекарств с образованием патологической популяции эритроцитов, у которых нарушена структура мембраны).
При наследственных гемолитических анемиях.отмечается усиленная регенерация эритроидного ростка, но эритропоэз часто может быть не эффективным (когда в костном мозге разрушаются ядерные формы эритроцитов). В мазке крови, наряду с регенеративными формами (высокий ретикулоцитоз, полихроматофилия, единичные ядерные формы эритроцитов),находится дегенеративно измененные клетки (например, микросфероциты при болезни Минковского-Шоффара).
При приобретенных гемолитических анемиях. степень уменьшения количества эритроцитов и гемоглобина зависит от интенсивности гемолиза. В мазках крови обнаруживают клетки физиологической регенерации и дегенеративно измененные эритроциты (пойкилоцитоз, анизоцитоз).
Приобретенная гемолитическая анемия может быть по типу кроветворения нормобластической, по регенераторной способности костного мозга - регенераторный, по ЦП - нормо - или гипохромной.
Анемии вследствие нарушения кровообразования.
B12-дефицитная и фолиеводефицитная анемия - это анемии связанные с нарушением синтеза нуклеиновых кислот и заменой нормобластического типа кроветворения мегалобластическим из-за недостатка в организме витамина B12 и фолиевой кислоты.
Причина:
1. Недостаток витамина в пище.
2. Неусвоение витамина B12 в желудке, что может быть связано с нарушением функции фундального отдела желудка, который вырабатывает гастромукопротеин (витамин B12 усваивается в комплексе с гастромукопртеином). Нарушение функции обкладочных клеток вызывается воздействием на них аутоантител (пернициозная или Аддисона-Бирмера или злокачественная анемия). Кроме того, подобное состояние может возникнуть после резекции желудка.
3. Неусвоение витамина B12 в кишечнике (при резекции тонкой кишки, опухоли, спру, дифиллоботриозе, алкоголизме).
4. Повышенное расходование витаминов при беременности.
5. Нарушение депонирования витаминов в печени при ее диффузном поражении.
Патогенез. Дефицит витамина B12 и фолиевой кислоты, участвующих в образовании тимина, входящего в состав ДНК, снижает скорость ее образования. Замедление репликации ДНК в тканях, где в норме деление клеток происходит наиболее интенсивно ( в кроветворной ткани) приводит к формированию крупных клеток крови: мегалоцитов, мегалобластов, гигантских мегакариоцитов. Созревание мегалобластов до мегалоцитов сопровождается нарушением энуклеации (об этом свидетельствуют появление в мегалоцитах телец Жолли (остатки ядра) и колец Кебота (остатки ядерной облочки)). Наличие большого количества мегалобластов и мегалоцитов, насыщенных гемоглобином, обуславливает гиперхромию (ЦП>1.0). В крови встречается много дегенеративно измененных эритроцитов(:пойкилоцитоз, анизоцитоз с микроцитозом, гиперхромия, мегалоциты с патологическими включениями). Уменьшается количество клеток физиологической регенерации (ретикулоциты, полихроматофилы), т.к. в костном мозге наблюдается раздражение эритроцитарного ростка с преобладанием мегалобластического типа кроветворения над нормобластическим. Наблюдается тромбо - и лейкоцитопения с атипическими клетками.
В результате недостатка витамина B12 в организме накапливается метилмалоновая кислота, которая токсична для нервных клеток. Кроме того, при дефиците витамина B12 в нервных волокнах синтезируются жирные кислоты с измененной структурой, что отражается на образовании миелина и приводит к повреждению аксона. Развивается дегенерация задних и боковых столбов спинного мозга (фуникулярный миелоз), поражаются черепно-мозговые и периферические нервы.
Дефицит витамина B 12 (цианкоболамина) приводит к:
1. Нарушению перехода: фолиевая кислота → тетрагидрофолиевая кислота → тимин → ДНК,. при котором страдают активно размножающиеся клетки кроветворной ткани (анемия);ЖКТ (воспалительно-атрофические процессы в слизистой).
2. Нарушение перехода метилмалоновой кислоты в янтарную (накопление метилмалоновой кислоты) оказывает токсическое действие на нервную систему.
3. Нарушение образования миелина в результате синтез жирных кислот с измененной структурой.
Железодефицитная анемия вызванна недостатком железа в организме в результате нарушения баланса между его поступлением, потреблением и потерей. Это самый распространненый вид анемии (80% всей заболевемости анемией).
Этиология.
1. Хронические кровопотери, приводящие к потере железа вместе с эритроцитами.
2. Повышенная потребность в железе (в период роста, созревания, беременности, лактации).
3. Алиментарная недостаточность железа.
4. Неусвоение железа:
а) при ахлоргидрии (соляная кислота ионизирует железо, что необходимо для его усвоения);
б) при авитаминозе витамина C (витамин C стабилизирует железо в двухвалентной форме, а трехвалетное железо организмом не усваевается);
в) при энтеритах и резекции тонкой кишки.
5. Нарушение транспорта железа (наследственная атрансферринемия, гипотрансферринемия при поражениях печени).
6. Недостаточная утилизация железа из его резерва (при инфекции, интоксикации).
7. Нарушение депонирования железа (при гепатитах, циррозах).
Недостаток железа в организме проявляется исчезновением гемосидерина в клетках печени и селезенки, снижением количества сидеробластов и сидероцитов в костном мозге. В крови уменьшается содержание сывороточного железа и степень насыщения им трансферрина (белка-переносчика железа), что ведет к снижению транспорта железа в костный мозг. Нарушается включение железа в эритроцитарные клетки, при этом снижается синтез гема и глобина, уменьшается активность некоторых ферментов в эритроцитах,что вызывает повышение их чувствительности к окислителям (т.к. неполноценность ферментативных процессов ведет к неустойчивости клеточных мембран), и эритроциты подвергаются гемолизу под действием окислителей. Продолжительность жизни эритроцитов сокращается.
Дефицит железа в организме приводит к уменьшению миоглобина и активности железосодержащих факторов тканевого дыхания. Развивается гемическая гипоксия, с атрофическими и дистрофическими процессами в тканях и органах (особенно в ЖКТ и миокарде).
Железодефицитная анемия - нормобластическая, гипохромная (из-за недостаточной гемоглобинизации). В мазке крови наблюдается анизоцитоз (микроцитоз), пойкилоцитоз. Количество ретикулоцитов зависит от регенерирующей способности костного мозга (анемия может быть в начале регенераторной, а затем гипорегенераторной).
ЛЕЙКОЦИТОЗЫ И ЛЕЙКОПЕНИИ
Патофизиология лейкоцитов очень важна в современной патологии, поскольку лейкоциты отражают внутреннее состояние организма и могут определять характер процесса, его тяжесть, прогноз и эффективность терапии.Они очень динамичны, очень быстро реагируют и очень информативны, определяются просто - поэтому их исследования обязательны в динамике.
Форма изменения:
1) общее число - могут быть лейкоцитозы выше 9000 в 1 микролитре и лейкопении - менее 4000 в 1 микролитре, но нужно иметь в виду, что даже у здоровых может быть порядка 2000 лейкоцитов в 1 мкл и реже 10000 лейкоцитов в 1 мкл.
2) изменение лейкоцитарной формулы - процентного соотношения лейкоцитов или изменения содержания отдельных видов лейкоцитов - это порциальные лейкоцитозы.
ЛЕЙКОЗЫ
Лейкозы (от греч. leuk - белый, относящийся к лейкоциту + os -патологический процесс, заболевание) — опухоли, возникающие из кроветворных клеток с первичным поражением костного мозга.
В 1845 Virchow описал у человека заболевание, которое характеризовалось резким увеличением числа белых кровяных телец, и дал этой болезни название "лейкемия" или "белокровие". В 1868-1878гг. Neumann установил, что при лейкемии первичные изменения возникают в костном мозге. В 1921г. Ellerman применил термин "лейкоз".
Сегодня известны несколько групп этиологических факторов, роль которых в возникновении лейкозов не вызывает сомнения:
Вирусы. К настоящему времени выделено и детально охарактеризовано
несколько типов вирусов, вызывающих различные виды лейкозов у животных.
Как правило, это РНК-содержащие вирусы, а также ДНК-содержащие вирусы,
которые относятся к герпес-вирусам. Вопрос о роли вирусов в происхождении
лейкозов у человека остается спорным.
Химические канцерогенные вещества. Лейкозы у работающих на
химических предприятиях и имеющих профессиональные контакты с бензолом
и продуктами его метаболизма описаны более 30 лет назад. Сейчас известны и
другие химические агенты, играющие существенную роль в их инициации:
химические растворители, инсектициды, нефтепродукты. Преимущественными
вариантами лейкозов, возникающих под действием химических канцерогенных
веществ, являются миеломоноцитарные, моноцитарные лейкозы, эритромиелоз
и хронический миелолейкоз (ХМЛ).
Ионизирующая радиация. Отмечено четырехкратное увеличение риска
заболеть лейкозом у взрослых, живших и работавших
километровой зоне вокруг АЭС. Величина риска
увеличивалась по мере приближения к станции и коррелировала со временем
проживания в зоне Установлено также увеличение заболеваемости
лейкозами среди жителей Южного Урала, подвергшихся воздействию продуктов
деления урана в результате аварии на радиохимическом предприятии "Маяк" в
1957-1958 гг.
Первые сообщения о росте числа лейкозов у пострадавших в Хиросиме и Нагасаки появились спустя полтора года после взрыва атомных бомб, первый пик заболеваемости зарегистрирован через 5-6 лет. В целом, рост заболеваемости лейкозами наблюдался в течение 40 лет после бомбардировки. Среди радиационно-индуцированных лейкозов преобладают хронические и острые миелоидные лейкозы (у взрослых), острые лимфобластные лейкозы (у детей).
Генная патология. В качестве возможных факторов наследственной
предрасположенности к лейкозам называют сниженную резистентность
хромосом к действию мутагенных агентов (феномен "хромосомной
нестабильности"), а также недостаточную активность ферментных систем
репаративного синтеза нуклеиновых кислот.
Согласно мутационно-клоновой теории, под действием канцерогенов возникает мутация в одной гемопоэтической клетке П-П1 классов. В результате изменяется пролиферация и дифференцировка мутировавшей клетки. Безудержное размножение этих клеток приводит к формированию клона "однотипных" опухолевых клеток. Это — моноклоновая стадия. В этот период опухолевые клетки чувствительны к химиотерапии. Важным условием, способствующим появлению мутировавших клеток, является снижение активности антимутационных механизмов противоопухолевой защиты.
В соответствии с современными представлениями злокачественная трансформация гемопоэтической клетки происходит в результате нарушения функции протоонкогенов и генов-супрессоров опухолевого роста. Эти гены в норме участвуют в регуляции клеточного цикла и дифференцировки. Их мутации могут сопровождаться повышением пролиферативного потенциала клетки.
Существуют две модели, которою описывают развитие хронических и острых лейкозов:
- Модель пошагового онкогенеза. Трансформация нормальной клетки в опухолевую происходит в результате единственной мутации "ключевого гена". Дополнительные мутации ("шаги") обеспечивают прогрессирование опухоли. Так развиваются хронические лейкозы.
- Модель "генетического груза". Развитию опухоли может предшествовать накопление в геноме некоторого количества мутаций. Это называется "генетическим грузом", который повышает способность клетки к пролиферации. "Генетический груз" может долгое время не проявляться вследствие функционирования генов-супрессоров опухолевого роста. Если же в последующем возникают мутации в генах-супрессорах, то клетка начинает интенсивно делиться. Появляется большое количество лейкозных клеток. Так развиваются острые лейкозы.
В процессе развития лейкоза происходят качественные изменения опухолевых клеток - формируется опухолевая прогрессия. К ее основным проявлениям относят следующие:
− трансформация лейкозов из моноклоновых в поликлоновые (новые клоны
появляются вследствие повторных мутаций);
− переход лейкозов от алейкемической формы к лейкемической;
− метастазирование лейкозных клеток в органы и ткани, которые в норме в
гемопоэзе не участвуют;
− угнетение нормальных ростков кроветворной ткани с развитием анемии,
тромбоцитопении, лейкопении;
− снижение числа зрелых лейкоцитов и увеличение количества властных
форм;
− уменьшение (утрата) ферментной специфичности лейкозных клеток;
− нарастание признаков клеточного атипизма;
− формирование устойчивости к воздействию противоопухолевых препаратов ускользание" лейкозов от лечения.
Процесс опухолевой прогрессии является основой формирования атипизма роста, обмена, функции и структуры лейкозных клеток. В костном мозге при лейкозах выявляются признаки патологического "омоложения" состава гемопоэтических клеток. Увеличивается число делящихся лейкозных клеток (в основном II-III классов), нарастает количество атипичных бластов. В основе этого явления лежит возрастание числа пролифилирующих лейкозных клеток, а также торможение или блокада процесса их созревания.
По морфологии клеток лейкозы делят на:
- острые, при которых основу опухоли составляют молодые, так называемые
властные клетки (клетки II, III, IV классов гемопоэза). Опухолевые клетки
утрачивают способность к дифференцировке.
- хронические, при которых субстрат опухоли составляют созревающие и зрелые клетки. Опухолевые клетки сохраняют способность к дифференцировке.
Властные клетки при остром лейкозе морфологически трудно различимы, но могут быть дифференцированы с помощью цитохимических методов. Исходя из этого было предложено подразделять острые лейкозы на острые лимфобластные и острые нелимфобластные.
В 1995 г. Европейской группой по иммунологической характеристике лейкозов была предложена иммунологическая классификация острых лимфобластных лейкозов (ОЛЛ). В соответствии с этой классификацией выделяют 4 варианта Т-ОЛЛ и 4 варианта В-ОЛЛ.
ПАТОФИЗИОЛОГИЯ ГЕМОСТАЗА
Гемостаз - остановка кровотечения - является гармоничным, локализованным и обратимым результатом нарушения равновесия в виде временной гиперкоагуляции.
Внутрисосудистое тромбообразование - тромбоз - это прижизненное свертывание крови в просвете сосуда.Тромбоз вызывает прижизненное расстройство кровообращения в тканях. В 50% случаев гибель человека связана с тромбозом сосудов. В основе патогенеза тромбоза лежат повреждения стенок сосудов, изменения функционального состояния системы гемостаза и замедление кровотока. Первопричиной тромбозов считают повреждение сосудистой стенки, на фоне которого происходит адгезия и агрегация тромбоцитов с образованием первичного тромба, а также активация синтеза простагландинов в тромбоцитах,свертывающей системе крови, локальная гиперкоагуляция, высвобождение фактора XIII и локальное торможение фибринолиза.
Повреждение стенок сосудов может быть структурным (в результате травмы, инфекций, реакции антиген-антитело и др.), и функциональным (снижение антитромбогенной активности сосудистой стенки при воздействии адреналина, норадреналина и кортизола при эмоциональном стрессе, а также активации перекисного окисления липидов при воздействии ионизирующего излучения, ожогах, атеросклерозе).
В патогенезе при структурных повреждениях создаются локальные условия для агрегации тромбоцитов и активации фактора XII. Однако при высокой скорости кровотока и антитромбогенной активности стенки сосуда, из которой постоянно в кровь выделяется простациклин, условия для возникновения тромбоза отсутствуют. При функциональном повреждении устраняется ингибирующее тромбогенез действие стенки сосуда, создаются условия для ускорения свертывания крови, спонтанной агрегации тромбоцитов и развивается тромбоз.
Тромбоз возникает чаще при нарушении биологической надежности системы гемостаза при повреждении ее регуляторных механизмов, ведущем к предтромбозному состоянию (тромбофилии). Тромбофилия возникает вследствие изменения одного или нескольких компонентов системы гемостаза, т.е. активации внешней и внутренней систем. Типичные примеры повышения активности факторов свертывания и обстоятельства их анормального присутствия в крови:
- тканевой тромбопластин (фактор III), поступающий из травмированных тканей, при рассасывании гематомы, введении в кровообращение жидкостей, обладающих высокой тромбопластической активностью (при акушерском вмешательстве, при резком гемолизе, спровоцированном микробными токсинами, изо- или аутоантителами, несовместимости крови.
- эндогенная активация XII фактора в сосудах, в отечных зонах эндотелия при травмах, продолжительном капиллярном стазе.
- увеличение числа тромбоцитов, усиление их элементарных свойств - агдгезии и агрегации.
Формы тромбоцитопатии:
Тромбоцитопении - как наиболее частые и яркие проявления спонтанных кровотечений. В норме 200-400 тыс/мкл, меньше 70000 - угроза кровотечений и меньше 30000 - критическая величина (в сутки 30000 тромбоцитов потребляется сосудистой стенкой).
Причины:
1. нарушение выработки тромбоцитов в костном мозге (болезнь Верльгофа) или эссенциальная тромбоцитопения при нарушении:
а) отшнуровывания тромбоцитов от мегакариоцитов, лейкозы, радиация, инфекции и интоксикации, злокачественные опухоли КМ, химиопрепараты - цитостатики, и
б) нарушение созревания мегакариоцитов в КМ.
2. гибель тромбоцитов в кровеносном русле при воздействии аутоантител, при инфекциях, интоксикациях, при повышении функции селезенки,
3. усиление потребления тромбоцитов при ДВС - тромбоцитопатия потребления.
Лабораторная диагностика:
1. с учетом того, что для патологии тромбоцитов характерны точечные кровоизлияния (петехии, а у детей - распространенные тромбоцитопенические пурпуры) - производят исследование костного мозга,
2. исследование содержания количества тромбоцитов в крови и тесты:
а) удлинение времени кровотечения,
б) снижение механической устойчивости капилляров (в связи с нарушением питания сосудистой стенки),
в) снижение ретракции сосудистого сгустка,
г) снижение адгезии и агрегации тромбоцитов.
Качественные изменения тромбоцитов:
1. нарушение способности тромбоцитов к адгезии,агрегации и выделению факторов свертывания крови (дистромбоцитозы при тромбоцитарных лейкозах)
2) недостаток 3 фактора - тромбопластического - тромбоцитодистрофия,
3) недостаток 6 фактора - ретрактозима - тромбоцитоастения.
Диагностика. Если количество тромбоцитов в норме, но восстановление свертываемости крови происходит при добавлении свежей крови - недостаток 3 фактора, а по ретракции сгустка судят о недостатке 6 фактора.
Коагулопатии.
В основу рабочей классификации может быть положена схема нормального свертывания крови. Тогда заболевания можно сгруппировать соответственно фазам свертывания крови:
● ГД (геморрагический диатез), обусловленные нарушением первой фазы свертывающей системы (дефицит факторов VIII, IX, XI и XII), наличием в крови ингибиторов к факторам VIII (гемофилия A) и IX (гемофилия B), дефицит тромбоцитарного компонента тромбопластинообразования, ангиогемофилия.
Гемофилия - характеризуется кровоточивостью крупных сосудов – кровоподтеки (90% у детей). Гемофилия передается женщинами, а проявляется у мужчин. У детей гемофилия проявляется в большей степени поражением сосудов, поскольку в них есть травматизация поверхностей, суставы распухают - анкилоз, сильные боли, ограничение движения → инвалидность. Патогенез - плохо активируются факторы свертывающей системы крови, или развивается их иммунное поражение.
Лабораторная диагностика - клинически - кровоподтеки,замедление свертывания крови, снижение тромбопластической активности крови.
● ГД, обусловленные нарушением второй фазы свертывающей системы крови: дефицит плазменных компонентов тромбинообразования - факторов II, V, VII и X при патологии печени, наличие антагонистов тромбинообразования (антитромбин I - фибрин, антитромбин II - гепарин, антитромбины III, IV, V, VI), наличие антагонистов к факторам протромбинового комплекса (II, V, VII, X).
Коагулопатия с поражением протромбинового комплекса напоминает гемофилию - гемофилоидные обширные кровоподтеки (при недостаточности витамина K, циррозах, желтухах), передозировке гепарина.
Лабораторная диагностика - снижение протромбинового индекса.
● ГД с нарушением III фазы. Причины:
а) нарушение образования фибриногена при патологии печени или РЭС,
б) усиленное потребление фибрина при тромбозе, ДВС-синдром,
в) патологическое усиление фибринолиза. Может быть врожденная недостаточность XIII фактора. Течение тяжелое.
Лабораторная диагностика - определение фибриногена и его фракций, уровня фибринолиза.
Тромбо-геморрагический синдром (ДВС) - неспецифический общепатологический процесс первоначальной гиперкоагуляции, связанный с поступлением в кровоток активаторов свертывания крови и агрегации тромбоцитов. Образуется тромбин и множество микросгустков и агрегатов клеток, блокирующих микроциркуляцию в органах. Наиболее частые причины:
1) тяжелая патология,
2) гипоксия тканей и клеток крови с активацией тканевого трмбопластина при гибели тканей,
3) травматизации,
4) иммунные повреждения тканей,
5) действии бактериальных токсинов,
6) при шоке,
7) ожеге,
8) распаде злокачественных опухолей,
9) массивном распаде эритроцитов и лейкоцитов,
10) тяжелой акушерской патологии.
Внутрисосудистое свертывание часто сочетается с активацией фибринолитической системы, расщеплением фибрина и фибриногена, высвобождением продуктов деградации фибрина (ПДФ). Этот процесс сопровождается сильной вазомоторной реакцией и не заканчивается до тех пор, пока коагуляционный механизм и вазомоторный аппарат не нормализуются и последние продукты деградации фибрина/фибриногена не будут удалены из крови.
Патогенез I фазы связан с резким повышением тромбопластической активности в сочетании с повреждением сосудистой стенки. Это приводит к диссеминированному тромбозу мелких и мельчайших сосудов, потреблению факторов свертывания крови - коагулопатия и тромбоцитопения потребления, потреблению естественного антикоагулянта крови - антитромбина III ..
Происходит истощение свертывающей, калликреин-кининовой и др. систем, гипоксия, ацидоз, дистрофия и глубокая дисфункция органов, интоксикация организма продуктами белкового распада и др. метаболитами тканей.
Активируются антикоагулянты и антиагреганты, активируется фибринолиз и происходит репаративное расплавление сгустков, направленное на восстановление проходимости сосудистых каналов. Однако активация фибринолиза принимает, как правило, генерализованный характер и лизируются не только микротромбы, но и повреждаются циркулирующие факторы свертывания крови, что еще больше усугубляет коагулопатию и развивается повторное кровотечение, остановить которое чрезвычайно сложно. Помимо прямого воздействия на свертывание крови, ПДФ блокируют сократительную способность гладких мышц.
Т.о. выделяют 2 фазы: гипер - и гипокоагуляции и соответственно фазам строится лабораторная диагностика:
В I фазу выявляется ускорение свертывания крови, повышение тромбопластической активности, а во II фазу - резкое снижение факторов II и III фазы вплоть до афибриногенемии (коагулопатия потребления).
В клинической практике различают острую, подострую и хроническую форму.
К острой и подострой формам можно отнести:
● геморрагические проявления - петехиальные кровоизлияния в месте иньекций, в склеру глаз, слизистую оболочку желудочно-кишечного тракта, профузные кровотечения из матки;
● тромботические проявления - некроз кожи в области кончика носа и мочки уха, внезапная ишемия конечностей, постинфарктные пневмонии, тромбозы магистральных сосудов;
● нарушения функции почек (олигоурия и анурия);
● нарушение функции ЦНС (эйфория, отсутствие критической оценки окружающего, дезориентация и оглушенность вплоть до комы);
● нарушения внешнего дыхания (одышка, акроцианоз, снижение артериализации крови и артериовенозной разницы по кислороду);
● внутрисосудистый гемолиз.
При острой форме синдрома ДВС на первое место выходят явления гипотонии, выраженной кровоточивости, шока, а при хронической - нарушения функции отдельных органов.
В клинике возможна экспресс диагностика нарушения коагуляции:
1. Время свертывания по Ли-Уайту - 1 мл венозной крови помещают в пробирку и каждые 30" пробирку наклоняют в одну и ту же сторону. На секундомере отмечают время образования полноценного сгустка:в норме у доноров 6.30"-10 мин; у рожениц 5-6 мин.
2. Метод определения фибриногена в крови по Мочабели: в пробирку помещают несколько мл венозной крови и:
а) уровень фибриногена в пределах нормы, если свернувшаяся кровь (уровень эритроцитов и плазмы) при наклоне пробирки не меняет форму;
б) уровень фибриногена упал - при наклоне пробирки плазма сохраняет горизонтальное положение, эритроциты не изменились;
в) фибриногена нет, поверхности эритроцитов и плазмы при наклоне пробирки сохраняют горизонтальное положение.
Открыт ДВС в 1957г. Марией Семеновной Мочабели.
ПАТОФИЗИОЛОГИЯ ПЕЧЕНИ
Печеночная недостаточность (ПН) характеризуется снижением одной, нескольких или всех функций печени ниже уровня, необходимого для обеспечения нормальной жизнедеятельности организма. ПН делится на виды по следующим признакам:
1) по числу нарушенных функций - на парциальную и тотальную,
2) по течению - на острую и хроническую,
3) по исходу - летальную и нелетальную.
Причины печеночной недостаточности. Можно выделить две группы причин печеночной недостаточности.
К первой группе относятся патологические процессы, локализующиеся в печени и в желчевыделительных путях, а именно:
а) гепатиты - вирусные, бактериальные, токсогенные;
б) дистрофии (гепатозы);
в) циррозы;
г) опухоли печени;
д) паразитарные поражения ее;
е) генетические дефекты гепатоцитов;
ж) камни, опухоли, воспаления желчевыделительных путей с выраженным холестазом.
Ко второй группе причин относятся патологические процессы вне печени, а именно:
а) шок, в том числе послеоперационный;
б) сердечная недостаточность;
в) общая гипоксия;
г) почечная недостаточность;
д) белковое голодание;
е) гипоавитаминоз E;
ж) дефицит селена;
з) эндокринопатии - в частности, острая недостаточность надпочечников;
и) метастазы опухолей в печень.
Патогенез печеночной недостаточности. Общий патогенез ПН может быть представлен в виде следующей цепи изменений: действие повреждающего фактора → 1) изменение молекулярной архитектоники мембран гепатоцитов → 2) усиление свободнорадикального перекисного окисления липидов (ПОЛ) → 3) частичная или полная деструкция мембран + повышение их проницаемости → 4) выход из лизосом их гидролаз, что потенцирует повреждение мембран клеток → 5) освобождение поврежденными макрофагами некрозогенного фактора и интерлейкина 1, способствующих развитию воспалительной и иммунной реакции в печени → 6) образование аутоантител и аутосенсибилизированных T-киллеров, вызывающих дополнительное аутоаллергическое повреждение гепатоцитов.
Каждое из перечисленных патогенетических звеньев может стать на определенной стадии развития печеночной недостаточности доминирующим, что должно быть учтено при выборе ее терапии.
ПАТОФИЗИОЛОГИЯ ПОЧЕК
Главным экскреторным органом являются почки. Они способны выделять из крови в мочу:
1) воду,
2) продукты обмена веществ, особенно белкового - мочевину и мочевую кислоту,
3) различные соли (т.е. почки - главный регулятор содержания в организме NaCl),
4) чужеродные вещества - яды, токсины микробов, ядовитые продукты, всосавшиеся из кишечника и обезвреженные печенью - индол - индиканскатол.
Почки тем самым поддерживают:
1) гомеостаз крови,
2) осмотического и онкотического давления крови,
3) кислотно-щелочного равновесия
По составу мочи можно судить не только о функции почек, но и о состоянии других органов и систем.
В настоящее время почка рассматривается как часть функциональной системы выделения. Как любая система она имеет:
I Афферентное звено, представленное осморецепторами тканей и волюморецепторами в устьях полых вен и предсердий,
Центр мочеотделения (открытый еще основоположником экспериментальной физиологии и патологии французским физиологом Клодом Бернаром, который установил, что укол в дно IV желудочка между ядрами слухового и блуждающего нервов вызывает полиурию). Полиурия может быть вызвана и уколом в серый бугор (tuber cinereum).
Эффекторным и рабочим органом системы являются почки. Почка очень богата как афферентными, так и эфферентными нервными волокнами. Они проходят главным образом по сосудам почек, образуя околопочечные нервные сплетения. Очень хорошо развита гуморальная регуляция.
Нарушенние мочеотделения может быть связано с поражением различных отделов этой системы.
Функцинальной единицей в почках является нефрон. В его состав входит:
1. Vena afferens - сосуд, приносящий кровь, в нем хорошо развит мышечный слой, регулирующий просвет сосуда.
2. Мальпигиев клубочек (rete mirabilis - чудесная сеть).
3. Vena efferens - сосуд, уносящий кровь от клубочка,
4. клубочек окружен и охвачен двухслойной капсулой Шумлянского-Боумена, имеющей висцеральный и париетальный листок.
5. Полость капсулы переходит в канальцевую систему нефрона, в которой различают 3 части проксимальная часть или извитые канальцы первого порядка, тонкий нисходящий и восходящий отдел или колено петли Генле, спускающийся из коркового вещества почки в мозговое и дистальная часть канальцевой системы - извитые канальцы II порядка, которые переходят в собирательные трубки, по которым образующаяся моча отводится в почечные лоханки, из них по мочеточникам в мочевой пузырь и по мочеиспускательному каналу - наружу.
Канальцы почек обладают секреторной функцией - почки активно секретируют антибиотики (пенициллин), йодосодержащие контрастные препараты, ионы калия, избыток его выделения ведет к калиевому диабету и нарушению работы сердца.
Почки обладают инкреторной функцией - выроботка ренина в юкстагломерулярном аппарате - ренин обеспечивает процессы саморегуляции мочевыделения.
Нарушение мочевыделения может проявляться в патологии механизмов образования мочи - фильтрации и реабсорбции. В настояшее время общепризнанной является фильтрационно-реабсорбционная теория мочеобразования - теория Ричардсона. Сущность теории Ричардсона - в начале фильтрации, затем обратная рабсорбция веществ в кровь или их концентрация в моче и выделение из организма.
Механизмы фильтрации обусловлены тем, что диаметр артериолы, приносящей кровь в 2 раза больше артериолы, выносящей кровь из клубочка. Благодаря этому в капиллярах клубочков возникает самое высокое давление. 60-80 мм ртутного столба вместо 30-40 мм ртутного столба в других тканях. Эта разница капиллярного давления создает гидростатическое давление, обуславливающее переход жидкой части крови из капилляров клубочка в капсулу Шумлянского. Величина этого давления зависит от разницы между гидростатическим давлением крови в капиллярах клубочков и величиной коллоидно-осмотического давления плазмы, которое удерживает воду и противостоит гидростатическому давлению. Оно равно 25-30 мм. ртутного столба. Кроме того, гидростатическому давлению противостоит внутрипочечное давление - это давление в почечных канальцах,обусловленное находящейся в них мочой - 8-10 мм ртутного столба.Таким образом фильтрационное давление ФД = гидростатическое давление - коллоидноосмотическое давление и внутрипочечное давление = 80-(30+10) = 40 мм ртутного столба. Вот почему падение давления крови до 40-50 мм ртутного столба является критическим и ведет к полному прекращению мочеотделения - анурии. Фильтрация осуществляется через стенку капилляра и висцеральный листок капсулы Шумлянского-Боумена. Толщина стенки, отделяющей ток крови от полости капсулы не превышает 1 мм. Фильтрующаяся жидкая часть крови очень близка по своему составу к плазме и называется провизорной или первичной мочой. В сутки в норме через почку проходит 500-600 литров крови и образуется 90-100 литров фильтрата - суточный диурез. Мочи же выделяется в норме 1.0-1.5 литра. В процессе прохождения фильтрата через канальцы и петлю Генле происходит обратная реабсорбция полностью или частично.
Некоторые авторы считают, что эпителий канальцев способен избирательно реабсорбировать различные вещества из первичной мочи в кровь. Считают, что эта избирательность определяется специфичностью функции клеток эпителия канальцев, что одни клетки реабсорбируют только глюкозу, другие только натрий и т.д. Все вещества по способности почек к их выделению делятся на 3 группы:
I. высокопроговые - глюкоза, магний, кальций. Эти вещества при нормальной их концентрации в плазме крови полностью или почти полностью реабсорбируются обратно в кровь, а с мочой они выделяются при повышении порога концентрации - например для глюкозы порог - 180 мг% в плазме.
II. низкопороговые вещества - мочевина, мочевая кислота и фосфаты - это такие вещества, часть которых реабсорбируется обратно, а часть выделяется с мочой. Полагают, что их обратная реабсорбция происходит в виде неактивной, а простой диффузии. Особенно обратной диффузии подвергается мочевина до 7 гр. на 90 литров.
III. Беспороговые вещества (сульфаты и креатин) совершенно не подвергаются реабсорбции и сколько бы не было в плазме - все они выделяются с мочой.
Для оценки фильтрационной, реабсорбционной и концентрационной функции почек определяются следующие показатели:
1) Величина диуреза в дневное и ночное время и
2) ее удельный вес 1018-1036,
3) наличие примесей белка, эритроцитов и
4) коэффициент очищения или клиренс, содержание хлоридов и мочевины.
В норме ночной диурез в 2-3 раза меньше дневного. При сердечной недостаточности, нефрите, нефросклерозе ночной диурез возрастает ( никтурия).
Клиренсом почек называется количество плазмы в крови , которое фильтруется через почки и очищается от определенного вещества в еденицу времени (минуту). Для определения клиренса обычно применяется гипуран, инулин, креатинин, сергозин. Все они фильтруются клубочками, а обратно канальцами не всасываются - т.е. это беспороговые вещества. Поэтому для определения функциональной способности канльцев применяются: фенолрот, диотраст, парааминогиппуровая кислота и другие вещества. Эти вещества выделяются преимущественно только канальцами. Принцип определения клиренса: утром натощак исследуемый выпивает 1 л жидкости, можно с бутербродом, затем в/м вводится фенолрот в виде раствора в количестве 25 мл. Через 10 минут после введения краски исследуемый должен опорожнить мочевой пузырь. Затем через каждые 20 минут в течении часа 3 раза собирают мочу и одновременно берут из вены кровь. Определяют концентрацию фенолрота в порциях крови и мочи, сравнивая их, делают расчет. В норме величина почечного плазмотока, который почка способна очистить в 1 минуту по фенолроту 400 см3, по диатрасту 700 мл плазмы. Основной причиной нарушения почек является воспалительные и дегенеративные склеротические процессы в самой почечной ткани.
Виды, этиология и патогенез нарушения мочевыделения. Нарушение мочевыделения может быть обусловлено:
1) нарушением мочеобразования, связанным с поражением почек,
2) нарушением мочеотделения так же в связи с поражение почек,
3) нарушение мочевыделения в результате поражения мочевыводящих путей.
Проявление нарушения функции почек может быть в двух формах -. изменение состава мочи и изменение диуреза.
Изменения состава мочи:
а) уменьшение в моче тех веществ, которые выделяются в норме - например уменьшение выделения мочевины,
б) появление в моче таких веществ, которые в норме не выделяются или появляются лишь в виде следов: лейкоциты, белки → альбуминурия - цилиндрурия, сахар → глюкозурия, крови → гематурия, биллирубинурия.
Изменения диуреза:
а) полиурия,
б) олигурия,
в) анурия.
Причины ренальные и экстраренальные.
Полиурия может быть вызвана повышенным введением в организм жидкости, особенно без содержания хлорида натрия
Регулируется этот процесс нейрогуморальным путем: повышение массы крови вызывает раздражение барорецепторов, которое ведет к угнетению выроботки альдостерона усиленному выделению почками натрий-хлора и снижению его в крови снижению осмотического давления крови изменение импульсации с осморецепторов, уменьшение обратной реабсорбции воды - полиурии и уменьшении массы крови.
Причины и механизмы полиурии ренального происхождения: наблюдается во второй, так называемой компенсированной, фазе хронической почечной недостаточности. При этом в результате поражения клубочков снижается их концентрационная способность - гипо- и изостенурия. В крови задерживается мочевина и другие вещества азотистого обмена. Попадая в почки в повышенной концентрации, они (как физиологические мочегонные вещества) вызывают ренальную полиурию. В первый период благодаря этому усилению мочеотделения почки компенсируют свою недостаточность концентрационной способности - т.к. больше выделяется и азотистых веществ. При этом полиурия может быть только в ночное время – так называемая никтурия. Никтурия является ранним признаком:
1) хронической сердечной недостаточности,
2) нефросклероза и
3) гломерулонефрита.
От полиурии следует отличать поллакиурию - учащение мочеиспускания при нормальном или повышенном диурезе (до 15-20 раз в сутки). Причиной поллакиурии является:
1) повышение чувствительности рецепторов мочевого пузыря, с которого - рефлекс на мочеиспускание, когда небольшое его наполнение вызывает рефлекс опорожнения. Причины:
а) воспаление-циститы,
б) опухолевые процессы,
в) камни,
г) пожилой возраст, сужение мочеиспускательного канала при гипертрофии предстательной железы, т.к. при этом мочеиспускание затруднено, то возникает мучительное состояние, желание и невозможность опорожнить мочевой пузырь - делают катетеризацию.
Одним из наиболее тяжелых последствий различных поражений почек является почечная недостаточность (ПН) - неспособность почек очищать кровь от продуктов обмена и поддерживать постоянство состава плазмы крови. По механизму возникновения и течения ПН может быть острая (ОПН) или хроническая (ХПН).
Виды, этиология и патогенез ОПН - внезапное нарушение функции почек вследствии действия на них экзогенных или эндогенных повреждающих факторов. Этиологические факторы ОПН делятся на 4 вида:
1) преренальные,
2) ренальные,
3) постренальные и
4) аренальные.
Преренальные причины ОПН:
а) внезапное падение АД (кровотечения, травма, шок, коллапс),
б) потеря больших колличеств воды и электролитов при неукротимой рвоте, поносе, передозировке мочегонных веществ. Все это приводит к уменьшению ОЦК, падению почечного плазмотока, снижению клубочковой фильтрации, - развитию первой, начальной стадии ОПН - олигоанурической. Таким образом, основным в патогенезе первоначальной ОПН является нарушение кровообращения в почках и ишемическое повреждение почечной ткани - гипоксия и даже некроз тканевых элементов.
ОПН ренального происхождения развивается как следствие прямого поражения паренхимы почек при отравлении специфическими нефротоксическими веществами, особенно солями: ртути, урана, хрома, отравление фосфором, уксусной кислотой, грибами, лекарственными препаратами - сульфаниламиды, антибиотики, вещества хининового ряда при передозировке или в результате повышенной индивидуальной чуствительности к препаратам. Механизм их действия - они оказывают прямое токсическое действие на эпителиальные клетки канальцев почек, вызывают
1) некробиотические изменения в протоплазме клеток эпитилия канальцев с последующим разрывом их базальной мембраны,
2) тубулорексис.
Анурия при этих измерения в почках обусловлена тем,что клубочковый фильтрат полностью
1) реабсорбируется в канальцах обратно или
2) уходит в межуточную ткань почки, а затем по межлимфотическим и венозным сосудам покидает почечную паренхиму, поступают в кровь и вызывают интоксикацию организма.
Причиной ренальной ОПН могут быть тяжелые токсико-аллергические процессы.
ОПН постренального происхождения - является следствием окклюзии (закупорки) верхних мочевыводящих путей камнями при мочекаменной болезни и опухолями различного происхождения.
Аренальная форма ОПН развивается в случае травматического размозжения обеих почек или удаления почек по жизненным показаниям.
Патогенез ОПН - острая блокада функций почек в первую очередь ведет к нарушениям внеклеточного гемостаза. Скопление различных веществ во внеклеточном пространстве создает повышенную нагрузку внутри клеток, где начинают преобладать катаболические процессы.
В течении ОПН выделяются 4 основные стадии:
1) начальная,
2) олигурическая,
3) полиурическая,
4) восстановительная или стадия выздоровления.
Начальная стадия ОПН обычно совпадает с периодом воздействия этиологического фактора (шок, коллапс, сепсис, отравление). Уже в первые сутки снижается диурез и развивается олигурия, наступает задержка жидкости и появляется гиперазотемия до 1.5-2 г/л.
В олигоанурической стадии (основная стадия болезни) - происходят наиболее тяжелые изменения гомеостаза и развертывается вся патогенетическая цепь: гипергидратация (задержка воды в тканях); гиперкалиемия (из-за выхода из клеток большого количества калия, который в условиях ацидоза может вызвать остановку сердца - это является наиболее частой причиной смерти при ОПН в стадии олигоанурии. Повышение в крови фосфатов ведет к гипокалциемии и склонностям к судорогам от недостатка кальция. Острую блокаду почек частично компенсируют в первой фазе другие органы - особенно ЖКТ, который за сутки выделяет до 3-4 гр. сухой мочевины. Мочевина частично выделяется и через кожу - потовые железы. Нередко на высоте уремической интоксикации на крыльях носа можно видеть иней - кристаллы мочевины. Однако компенсаторные возможности очень ограничены. Поэтому обычно довольно быстро развивается крайне тяжелое состояние и при отсутствии надлежащего лечения (особенно присоединения к искусственной почке) имеет место высокая летальность. Кроме того развиваются гиперазотемия (распад тканевых белков; гипонатриемия как результат разведения крови, т.е. гипергидратации крови. Очень быстро нарушается эритропоэз → анемия (Hb до 50-60 г/л). Если же острая стадия проходит, то наступает полиурия и через несколько месяцев почечная функция восстанавливается.
Полиуричесская стадия протекает в 2 этапа. Первый этап - ранняя диуретическая фаза, второй - фаза полиурии. Диурез нарастает постепенно в течении 4-5 суток, количество мочи увеличивается с 400-500 мл до 2-4 л. Вначале моча имеет низкий удельный вес и пониженное содержание мочевины и креатина. Снижение концентрационной способности почек сохраняется 2-3 недели и затем восстанавливается. Более длительное время держится анемия.
Для полиурической стадии характерно:
1) гипогидратация (выделение воды);
2) гипокалиемия,
3) гипонатриемия,
4) гипокальциемия.
Стадия выздоровления или восстановительная продолжается в течение нескольких месяцев в зависимости от тяжести и продолжительности ОПН.
Этиология и патогенез хронической почечной недостаточности. ХПН рассматривается как осложнение прогрессирующих заболеваний почек или единственной почки, а так же может быть как самостоятельное патологическое состояние, которое требует особых специфических форм лечения.
Этиология ХПН. К развитию ХПН от ее начальной до терминальной стадии могут привести следующие заболевания почек и мочеполовых путей:
1. первичные поражения клубочков - хронический гломерулонефрит до 30%, гломерулосклероз, вторичносморщенная почка,
2. первичные поражения канальцев (до 30%): наследственные заболевания, например, наследственный дефект ферментных систем канальцев (синдром Фанкони); хроническое отравление солями тяжелых металлов - свинец, кадмий, ртуть; хроническая идиопатическая гиперкальциемия,
3. сосудистые заболевания, ведущие к двустороннему первичному нефросклерозу или первичносморщенной почке; злокачественная эссенциальная гипертония, двухсторонний стеноз почечных артерий,
4. инфекционные заболевания почек, хронический пиелонефрит, ТВС до 30%,
5. обструктивные заболевания мочевых путей - верхних - камни, опухоли и нижних - аномалии развития шейки мочевого пузыря, аденома предстательной железы, структура мочеиспускательного канала,
6. коллагеновые заболевания:
а) склеродермия,
б) диссеминированная красная волчанка,
в) узелковый периартериит,
7. обменные заболевания почек:
а) амилоидоз,
б) подагра с мочекислой нефропатией,
в) первичный гиперпаратиреоидизм с гиперкальциемией,
8. врожденные двухсторонние аномалии почек и мочеточников:
а) двухсторонняя гипоплазия,
б) губчатая почка,
в) поликистоз почек,
г) нервно-мышечная дисплазия мочеточников,
9. радиационный интерстициальный нефрит.
В течении ХПН выделяют 4 стадии:
I . Латентная ХПН характеризуется скудностью субъективных и объективных симптомов и выявляются лишь при всестороннем обследовании. Может быть нарушение способности к концентрации мочи и явления гипо- и изостенурии. Клубочковая фильтрация остается нормальной или сниженной незначительно (до 50-60 мл/мин). Может быть
1) протеинтурия,
2) дисаминоацидурия (появление в моче аминокислот),
3) увеличение экскреции сахаров - глюкозоурия,
4) увеличение клиренса по гипурану.
II . Компенсированная ХПН наступает при более значительном снижении функции почек. При этом:
1) повышения содержания в крови мочевины и креатина еще нет,
2) суточный диурез,как правило, возрастает до 2-2.5 л за счет уменьшения канальцевой реабсорбии,
3) клубочковая фильтрация снижается более значительно (до 50-30 мл/мин.),
4) снижается осмолярность - т.е. осмотическое давление мочи,
5) могут возникать электролитные сдвиги - т.е.может быть полиурия, гематурия, цилиндрурия, бактереурия.
III . Интермитирующая стадия ХПН - характеризуется переходом от компенсации к декомпенсации и проявляется дальнейшим снижением клубочковой фильтрации и канальцевой реабсорбиции. В крови периодически появляется гиперазотемия - до 0.8 г/л мочевины и до 0.04-0.05 г/л креатинина. Клубочковая фильтрация снижается уже до 25 мл/мин. Для этой стадии ХПН характерна смена периодов улучшения состояния и ухудшения больного. Причинами обострения могут быть пиелонефрит, различные интеркурентные заболевания.
IV . При отсутствии необходимого лечения болезнь переходит в следующую стадию - терминальную или декомпенсации - эта стадия является необратимой, т.к. погибшие нефроны не способны к регенерации. Она сопровождается олигурией, анурией, уремией и уремической комой.
Патогенез почечных отеков.
При поражении почек могут возникать отеки:
1) нефротические (при нефрозах т.е. поражении канальцев) и
2) нефритические (при нефритах - поражении клубочков почек).
Патогенез отеков при нефрозах - поражение преимущественно канальцевого аппарата почек → увеличение проницаемости почечного фильтра для белков - альбуминурия → гипоальбуминемия → снижение онкотического давления крови → увеличение оттока воды в ткани - недостаточность обратного тока лимфы → уменьшение объема плазмы → гиповолемия → увеличение образования альдостерона и АДГ → задержка в организме натрия и воды → отеки по утрам на лице (особенно веки - где самая тонкая кожа).
Патогенез нефритических отеков связан с поражением клубочков - ведет к нарушению в них кровообращения, увеличению выработки ренина, который увеличивает образование ангиотензина- I и II, который активирует секрецию альдостерона. Альдостерон вызывает задержку натрия и воды → гипернатриемия - через осморецепторы активирует секрецию АДГ. АДГ активирует гиалуронидазу эпителия почечных и собирательных канальцев, разрушающую гиалуроновую кислоту стенки капилляров, повышая их проницаемость. Возникает генерализованный капаллярит - резко повышается обратная реабсорбация, вода задерживается в организме, а повышение проницаемости капилляров ведет к поступлению воды в ткани и возникновению отека. При этом в ткани выходит не только вода, но и белки плазмы крови. Поэтому отличительной чертой нефритических отеков является высокое содержание белка в межтканевой жидкости и повышение гидрофильности тканей. Отекам способствует также задержка натрия в тканях и повышение в них осмотического давления.
Патогенез уремии - задержка в организме всех тех ядовитых продуктов (особенно белков) которые в норме выделяются из организма с мочой, т.е. в крови накапливаются составные части мочи: увеличение
1) остаточного азота крови с 20-40 мг% до 500-700 мг%,
2) мочевины с 15-25 мг% до 400-500 мг%,
3) мочевой кислоты с 2-4 мг% до 10-20 мг%,
4) креатина с 1-1.5 мг% до 30-35 мг%,
5) индикана с 0.001 мг% до 6-7 мг% (т.е. в 6000-7000 раз).
Происходит отравление организма и нарастающие явления интоксикации. Полагают, что отравление вызывается не самой мочевиной, а углекислым и карбаминовокислым аммонием. Поскольку мочевина в большом количестве выделяется в кишечнике, то под влиянием бактерий кишечника она превращается в токсическую форму углекислый и карбаминовокислый аммоний, который всасываясь из кишечника – отравляет организм. Большое значение в механизмах интоксикации при уремии имеет накопление в крови фенольных соединений: фенола, крезола, индолуксусной и других кислот.Что такое уремия - сложный симптомокомплекс явлений самоотравления организма продуктами азотистого обмена, мочевины, мочевой кислоты и других веществ, накапливающихся в организме. Это финал прекращения фильтрационной и концентрационной способности почек.
1. Интоксикация при уремии характеризуется определенными явлениями со стороны ЦСН: постоянные, упорные, резкие не прекращающиеся днем и ночью головные боли как следствие влияния токсических веществ, нарушение обмена кининов и возникновение отека мозга с определенными симптомами: сонливость, бред, галлюцинации, снижение слуха и зрения → потеря сознания - уремическая кома.
2. Раздражение продуктами азотистого обмена → упорные, очень мучительные рвоты (мучительные - центрального происхождения на пустой желудок на фоне отвращения к мясному и потери аппетита). При рвоте пустой желудок как бы выворачивает наизнанку, не принося облегчения. Развивается уремические гастрит, бронхит, изо рта пахнет мочой (foetor uraemicus).
3. На коже выделяется в виде соли мочевина.
4. Интоксикация ДЦ ведет к дыханию Чейн-Стокса.
Нефротический синдром. Почечная недостаточность может быть следствием так называемого нефротического синдрома, который может быть следствием:
1) первичных заболеваний почек: гломерулонефрит - воспаление клубочков почек, амилоидоз -дистрофические изменения в канальцах, острый и хронический пиелонефрит, опухоли почек, нефропатии беременных.
2) вторичные заболевания - сифилис, системная красная волчанка, ожоговая болезнь, сахарный диабет, капиллярный гломерулонефрит или болезньнь Киммельстиля-Уилсона, заболевания крови - лейкозы, проявляется нефротический синдром массивной протеинурией и как следствие этого - гипопротеинемия → снижение онкотического давления крови, выход жидкости в ткани → развитие отеков.
Механизмы фильтрации.В настоящее время установлено,что фильтрация и гидростатическое давление активно регулируется юкстагломерулярным аппаратом (ЮГА), открытым учеными Юка и Пикелинг. Клетки этого аппарата являются 1) рецепторами ЮГА и 2) его эффекторами. Они обладают способностью выделять ренин. Количество выделяемого ренина зависит от натяжения мембраны оболочки клетки, а это зависит от кровяного давления в клубочках почек. Ренин сам не активен, он влияет на α2-глобулин (ангиотензиноген) - отщепляет пептид из 13 аминокислот – ангиотензин-I, который под действием находящейся в крови дипептидкарбоксипептидазы превращается в ангиотензин II (отщепляется еще 3 аминокислоты).
Установлено, что ренин выделяется в виде гранул при напряжении мембраны клеточной оболочки вследствие падения давления крови в клубочках. У человека при поражении задней доли гипофиза и прекращении выделения АДГ диурез может достигать теоретически максимальных величин. Это объясняется тем, что АДГ способствует обратному всасыванию воды из канальцев в кровь. В отсутствие этого гормона совершенно прекращается активная реабсорбция воды в дистальном сегменте петли Генле - диурез = 17 см3/мин = 25 л/сутки. Возникает несахарный диабет.
АКРОМЕГАЛИЯ
Акромегалия - заболевание, обусловленное избыточной секрецией гормона роста, сопровождающейся диспропорциональным ростом костей, мягких тканей, внутренних органов. В детском и юношеском возрасте (при незавершенном окостенении гипофизарных хрящей и сохранении зон роста) отмечается относительно пропорциональный рост скелета с развитием гигантизма. Принято расценивать рост выше 200 см как гигантизм, 180-200 см как субгигантизм.
Этиология и патогенез. Наиболее часто источником гиперпродукции соматотропного гормона (СТГ) является эозинофильная аденома гипофиза. Описаны случаи развития акромегалии при хромофобной аденоме, а также опухолях различной локализации, секретирующих вещества с соматотропной активностью. У части больных имеет место гиперплазия эозинофильных клеток аденогипофиза под влиянием гиперсекреции соматотропин-рилизинг-гормона (СРГ) гипоталамусом.
Большие количества СТГ и соматомединов вызывают усиление синтеза белков, хондроитинсульфата и коллагена в тканях, что обуславливает рост хряща и увеличение паренхиматозных органов. Кроме того, СТГ стимулирует липолиз и снижает чувствительность тканей к инсулину (контринсулярный эффект), а также способствует задержке в организме натрия, хлора и фосфатов.
ГИПОФИЗАРНЫЙ НАНИЗМ
Гипофизарная карликовость, гипофизарная микросомия— заболевание, связанное с дефицитом СТГ, результатом чего является задержка роста и физического развития. При отсутствии лечения рост мужчин не превышает 130 см, женщин - - 120 см.
В большинстве случаев причину гипофизарного нанизма идентифицировать не удается. Существенное значение придается генетическому фактору, а также органическим поражениям ЦНС, возникшим внутриутробно или в первые годы жизни ребенка. Нарушение секреции СТГ может быть также результатом опухоли, воспалительного или токсического повреждения гипоталамуса с нарушением секреции рилизинг-гормонов (либеринов). Принято выделять абсолютную недостаточность СТГ, вызванную нарушением его секреции, и относительную, связанную со снижением тканевой чувствительности к этому гормону, а также с дефицитом в тканях инсулиноподобных факторов роста.
Недостаточность секреции СТГ очень редко бывает изолированной. Как правило, у больных гипофизарным нанизмом имеет место снижение других тропных гормонов с соответствующими клиническими проявлениями (гипотиреоз, гипогонадизм и др.). Ввиду того, что гипофизарный нанизм развивается в эмбриональном периоде или раннем детстве, заболевание проявляется резкой задержкой роста и физического развития.
НЕСАХАРНЫЙ ДИАБЕТ
Несахарный диабет заболевание, характеризующееся абсолютной или относительной недостаточностью антидиуретического гормона (АДГ), сопровождающейся нарушением реабсорбции воды в дистальных отделах извитых канальцев почек, развитием полиурии с низкой относительной плотностью мочи и полидипсии. Заболевание чаще развивается в молодом возрасте.
Этиология и патогенез. Заболевание вызывается патологическими процессами в диэнцефало—гипофизарной системе (повреждение нейросекреторных ядер переднего гипоталамуса - - супраоптических и паравентрикулярных, в которых вырабатывается АДГ, нейрогипофиза и соединяющих их нервных путей). Наиболее частыми причинами несахарного диабета являются инфекция (грипп, скарлатина, брюшной тиф, сифилис, послеродовой сепсис и др.), травма, объемный процесс. У части больных заболевание может быть вызвано инфильтрацией гипоталамо-гипофизарной системы патологическими элементами крови при гемобластозах. Довольно часто причину заболевания установить не удается - так называемый «идиопатический» несахарный диабет имеет обычно наследственную природу. В отдельных случаях может иметь место относительная недостаточность АДГ вследствие повышенной его инактивации на периферии или снижении чувствительности рецепторного аппарата дистальных отделов почечных канальцев. В любом случае дефицит АДГ (или его эффекта) сопровождается торможением обратного всасывания воды в дистальных отделах канальцев, развитием полиурии и жажды. Описаны семейные формы несахарного диабета, при которых нейроны гипоталамуса теряют способность синтезировать вазопрессин вследствие определенного генетического дефекта. Этиология почечной формы несахарного диабета изучена недостаточно.
ЗАБОЛЕВАНИЯ НАДПОЧЕЧНИКОВ
ФЕОХРОМОЦИТОМА
Собирательное понятие опухолей мозгового вещества надпочечников или вненадпочечниковой хромаффинной ткани - - хромаффинома, которая может быть доброкачественной (феохромоцитома), или, редко, злокачественной (феохромобластома). При обеих формах вырабатываются в избыточном количестве катехоламины. Феохромоцитома чаще всего локализуется в мозговом слое надпочечников, хотя формально возможна и внепочечная ее локализация.
Этиология заболевания неизвестна. В ряде случаев доказан семейно-на-следственный характер заболевания. Установлено, что наследственное происхождение опухоли определяется геном доминантного типа. Для семейных форм феохромоцитом характерно двустороннее поражение и более высокая частота малигнизации. Существует два вида опухоли: преимущественно продуцирующая адреналин и продуцирующая норадреналин.
Патогенез обусловлен выбросом катехоламинов и сводится к картине симпатико-адреналовых «бурь». Наряду с повышением АД выявляются вегетативные расстройства: тахикардия, сильная потливость, тошнота, рвота, нарушение зрения, тремор. Опухоль секретирует преимущественно адреналин, ДОФА и дофамин. Воздействие катехоламинов на сердце может проявляться гипертрофией, в некоторых случаях сердечной недостаточностью. Наиболее характерно наличие специфической кардиомиопатии, особенно у больных с частыми выбросами катехоламинов.
По характеру и частоте этих вегетативных пароксизмов рассматривают три варианта заболевания:
1) пароксизмальная форма повышения АД, при этом вне приступа давление снижается до нормальных величин;
2) так называемая постоянная форма (постоянно высокое АД), развивающаяся при наличии отягощенной по АГ наследственности;
3) смешанная форма, когда на фоне АГ у больного происходят как бы дополнительные гипертензивные кризы с вегетативными проявлениями.
ДИФФУЗНЫЙ ТОКСИЧЕСКИЙ ЗОБ
Аутоиммунное, генетически обусловленное заболевание, основными клиническими признаками которого являются увеличение щитовидной железы, экзофтальм и синдром тиреотоксикоза, возникающий в результате повышения секреции тиреоидных гормонов. По МКБ-10 (тиреотоксикоз).
Соотношение числа болеющих мужчин и женщин составляет 1:10.
Причины тиреотоксикоза:
I. Повышенная продукция тиреоидных гормонов щитовидной железы может быть:
ТТГ-независимой: при диффузном токсическом зобе - болезнь Грейвса—Базедова; функциональной автономии щитовидной железы; унифокальной функциональной автономии (в том числе тиреотоксическая аденома); мультифокальной функциональной автономии(в том числе многоузловой токсический зоб); диссеминированной функциональной автономии (диффузное распределение автономно функционирующих тиреоцитов в виде мелкоузловых ареалов); йод-индуцированного тиреотоксикоза (йод-Базедов); высоко- дифференцированном раке щитовидной железы; гестационном тиреотоксикозе; хорионкарциноме, пузырном заносе; аутосомно-доминантном неиммуногенном тиреотоксикозе.
ТТГ-зависимой: при ТТГ продуцирующей аденоме гипофиза (ти-
реотропинома); синдроме неадекватной секреции ТТГ (резистентность тире-отрофов к тиреоидным гормонам).
П. Повышенная продукция тиреоидных гормонов может быть ятрогенной и артифициальной.
III. Продукция тиреоидных гормонов вне щитовидной железы - при struma ovarii, функционально активных метастазах рака щитовидной железы.
В настоящее время основное значение в развитии заболевания придается генетически обусловленным изменениям в системе иммунного контроля с нарушением функции Т-супрессоров, пролиферацией запрещенных клонов лимфоцитов и их сенсибилизацией с цитотоксическим действием Т-лимфоцитов на клетки-мишени ЩЖ. Иммунокомпетентные Т-лимфоциты, реагируя с аутоантигенами ЩЖ, стимулируют образование аутоантител, которые, в свою очередь, наделены способностью вызывать гипертрофию ЩЖ и ее гиперфункцию. Для суммарного обозначения различных тиреостимулирующих факторов пользуются термином «тиреостимулирующие иммуноглобулины» (ТСИ). Все они являются иммуноглобулинами класса G, способны успешно конкурировать с тиреотропным гормоном (ТТГ) за места связывания с рецепторами ТТГ и тем самым оказывать стимулирующий эффект на ЩЖ. Уровень ТТГ при этом, как правило, понижается. Патогенез основных клинических проявлений болезни связан с влиянием избытка тиреоидных гормонов на основные органы и системы - - в первую очередь ЦНС, ССС и др. Часть симптомов обусловлена калоригенным эффектом тиреоидных гормонов, который проявляется стимуляцией тканевого дыхания, повышением основного обмена, ускорением метаболизма многих гормонов и др. Многообразие эффектов триеоидных гормонов объясняет разнообразие клинических проявлений заболевания.
ГИПОТИРЕОЗ
- Синдром патологического снижения функции щитовидной железы различной природы. По патогенетическому принципу принято выделять первичный гипотиреоз, вызванный патологическим процессом в щитовидной железы, вторичный гипотиреоз, обусловленный повреждением гипоталамо-гипофизарной системы и третичный, обусловленный снижением секреции ТРГ гипоталамусом. Тяжелые формы гипотиреоза называют иногда микседемой за наличие выраженного слизистого отека (экстрацеллюлярного накопления мукополисахаридов).
Причины первичного гипотиреоза многообразны;
- врожденная аплазия и гипоплазия ЩЖ, дефицит йода в окружающей среде, передозировка антитиреоидных препаратов, тиреоидэктомия, терапия радиоактивным йодом. Наиболее частая причина спонтанного гипотиреоза
- аутоиммунный тиреоидит (зоб Хашимото) заболевание с генетически
обусловленным дефектом иммунокомпетентных клеток. В крови больных при этом выявляются AT против различных компонентов ткани щитовидной железы
- тиреоглобулина, микросомальной фракции, рецептора ТТГ.
Щитовидная железа инфильтрируется лимфоцитами, плазматическими клетками. Деструкция фолликулярных клеток и фолликулов сочетается с признаками тиреоидной гиперплазии и разрастанием соединительной ткани. Выраженность пролиферативных и фиброзных изменений зависит от стадии патологического процесса. Исход аутоиммунного тиреоидита гипотиреоз. Снижение выработки тиреоидных гормонов сопровождается повышением концентрации ТТГ, что способствует гиперпластическим процессам в щитовидной железе, т.е. формированию зоба. В отдельных случаях роста зоба не происходит, несмотря на активную тиреотропную стимуляцию (атрофическая форма аутоиммунного тиреоидита).
Вторичный гипотиреоз является результатом поражения диэнцефало-гипофизарной системы (опухоли, травмы, нейроинфекции, сосудистые нарушения и др.), приводящего к снижению выработки ТТГ. Изолированное снижение уровня ТТГ наблюдается очень редко, чаще имеет место выпадение и других тройных функций аденогипофиза. Для вторичного гипотиреоза не характерно наличие зоба. Дефицит тиреоидных гормонов сопровождается угнетением всех видов обмена, снижением активности многих ферментных систем, функциональными нарушениями ЦНС, формированием своеобразного слизистого отека в различных органах и тканях с развитием дистрофических процессов. Особенно тяжелые нарушения наблюдаются при развитии гипотиреоза в раннем детском возрасте. У детей с врожденным гипотиреозом, не получавших лечения в первые годы жизни, отмечается необратимое отставание психического развития (кретинизм, идиотия), задержка физического и полового развития, резко нарушается формирование скелета, запаздывает появление центров окостенения, закрытие эпифизарных щелей, прорезывание зубов.
ПЕРВИЧНЫЙ ГИПЕРПАРАТИРЕОЗ
Гиперпаратиреоз представляет собой эндокринное заболевание, связанное с повышенной продукцией паратгормона. Оно известно под названиями: болезнь
Реклингаузена, генерализованная фиброзно-кистозная остеодистрофия, кистозная остеодистрофия, гиперпаратиреоидизма.
Заболеваемость гиперпаратиреозом составляет 1 случай на 100 000 жителей в год. Это болезнь среднего возраста (от 20 до 50 лет), женщины болеют в 2—3 раза чаще мужчин.
Источник гиперпродукции ПТГ - аденома (одна или несколько), реже - - аденокарцинома ОЩЖ. ПТГ - один из основных регуляторов фосфорно-кальциевого обмена, поэтому значительная часть симптомов гиперпаратиреоза обусловлена нарушением гомеостаза кальция и фосфора, в первую очередь - - гиперкальциемией. Гиперкальциемический эффект ПТГ связан с воздействием на органы-мишени: почки, кости и, в меньшей степени, кишечник. Почечный эффект характеризуется стимуляцией экскреции фосфора. Гипофосфатемия компенсируется мобилизацией фосфатов из костного депо, где они сосредоточены в соединении с кальцием. Следствием этих процессов является, с одной стороны,— развитие нефролитиаза, а с другой - - создаются условия для деминерализации скелета. Нарушение метаболизма в костной ткани усугубляется способностью ПТГ повышать количество и метаболическую активность остеокластов, стимулировать продукцию цитратов и лактата остеоцитами, подавлять синтез гликозоаминогликанов, которые являются существенным компонентом органической основы кости. Следствия гиперкальциемии - - снижение нервно-мышечной возбудимости, АГ, склонность к язвообразованию в ЖКТ, тромбоэмболическим осложнениям.
ГИПОПАРАТИРЕОЗ
Гипопаратиреоз (недостаточность ОЩЖ) - заболевание, характеризующееся недостаточной секрецией ПТГ, приводящей к нарушению фосфорно-кальциевого обмена.
Этиология и патогенез. Наиболее частая причина заболевания травма-тизация или удаление ОЩЖ при операции на ЩЖ. Определенное значение имеет лучевое повреждение ОЩЖ, токсические воздействия, инфекция, кан-дидомикоз. Нередко наблюдается так называемый идиопатический гипопаратиреоз, причина которого неясна. Предполагается наличие генетического фактора, аутоиммунных нарушений.
Патогенез заболевания связан с дефицитом ПТГ, приводящим к снижению экскреции фосфора с мочой, повышению его концентрации в крови. Одновременно снижается энтеральное всасывание кальция в кишечнике и его мобилизация из костного депо. На фоне гипопаратиреоза снижается образование активной формы витамина D в почках - главного стимулятора синтеза кальций-связывающего белка в слизистой оболочке тонкой кишки. Присутствие этого белка в 3-5 раз повышает эффективность энтерального всасывания кальция. Большинство симптомов гипопаратиреоза связаны с гипокальциемией. При недостатке кальция во внеклеточной жидкости изменяется проницаемость клеточных мембран, что сопровождается повышением нервно-мышечной возбудимости. Характерны трофические расстройства, особенно в тканях эктодермального происхождения.
Иногда нарушаются процессы свертывания крови, результатом чего может быть повышенная кровоточивость. Ввиду многообразия функций, присущих биологически активному элементу кальцию, многообразны и клинические проявления его дефицита.
ПЕРВИЧНЫЙ САХАРНЫЙ ДИАБЕТ
L Инсулинзависимый сахарный диабет (ИЗСД или I типа СД) - абсолютная недостаточность инсулина. Заболевание, протекающее с абсолютной инсулиновой недостаточностью, возникающей вследствие иммунопатологических процессов, развивающихся на фоне генетической, конституциональной предрасположенности. Для ИЗСД характерно начало болезни в детском и юношеском возрасте, выраженность клинической симптоматики, лабильность течения, склонность к кетоацидозу, частое развитие микроангиопатий. Заместительная инсулинотерапия в подавляющем большинстве случаев является единственным способом сохранения жизни больного.
Этиология и патогенез. ИЗСД - - аутоиммунное заболевание. В основе его развития лежит наследственный дефект в 6-1 хромосоме, связанный с системой HLA-ДЗ, Д4. Определенную роль отводят вирусам свинки, кори, Коксаки, тяжелым стрессовым ситуациям, химическим веществам. Многие вирусы обладают сходством с бетта-клетками ПЖ. Нормальная иммунная система противостоит вирусам. При генном дефекте происходит инфильтрация островков лимфоцитами. В-лимфоциты вырабатывают цитотоксические AT. Бетта-клетки гибнут, и развивается недостаточность выработки инсулина - - формируется СД. 2. Инсулиннезависимый сахарный диабет (ИНСД или II типа СД). Протекает с относительной недостаточностью инсулина. В крови у больных уровень инсулина в норме или повышен. Основные критерии ИНСД: зрелый и пожилой возраст больных, нередко повышенная масса тела, как правило, постепенное начало заболевания, стабильное течение, отсутствие кетоацидоза, частое сочетание с атеросклеротическим поражением крупных сосудов.
Этиология и патогенез. В настоящее время генетическая основа ИНСД не вызывает сомнений. Генетические детерминанты при ИНСД носят еще более важный характер, чем при СД 1 типа. Подтверждением генетической основы ИНСД служит факт, что у однояйцевых близнецов ИНСД развивается почти всегда (95-100%) у обоих. В то же время генетический дефект, определяющий развитие ИНСД, до конца не расшифрован. С позиций сегодняшнего дня рассматриваются два варианта. Первый: два независимых гена вовлечены в патогенез ИНСД, один отвечает за нарушение секреции инсулина, второй вызывает развитие инсулинорезистентности. Рассматривается также вариант наличия общего дефекта в системе узнавания глюкозы бетта-клетками или периферическими тканями, в результате или снижается транспорт глюкозы, или снижается глюкозо-стимулированный ответ бетта-клеток. Основной патогенетический фактор, обусловливающий развитие ИНСД - - относительная инсулиновая недостаточность вследствие тканевой инсулинорезистентности. Большинство больных с этим типом диабета не нуждается в заместительной инсулинотерапии и требует диетического лечения или использования таблетированных противодиабетических препаратов.
В основе ИНСД - - генетический дефект в самих бетта-клетках и периферических тканях, проявляется без действия внешних факторов. Уменьшается чувствительность периферических тканей к действию инсулина; изменяется структура инсулина. На ИНСД влияет ожирение. При этом клеткам требуется большее количество инсулина, но его рецепторов в клетках не хватает.
ОСТРЫЕ ОСЛОЖНЕНИЯ ДИАБЕТА
Диабетический кетоацидоз (ДКА)—клинико-биохимический синдром с высоким уровнем глюкозы в крови, глюкозурией, гиперкетонемией. ДКА -- острое очень тяжелое состояние, из которого самостоятельно больной выйти не может, смерть наступает в течение 3—4 дней. Смертность от ДКА - - 5-6%.
На фоне нарастания гипергликемии жажда и полиурия становятся мучительными, выраженный катаболизм белка и электролитные потери порождают резкую мышечную слабость; накопление недоокисленных продуктов обмена -ч (ацетоуксусной и бетта-оксимасляной кислот) вызывают ацидоз и явления интоксикации - - анорексию, тошноту, рвоту, что в свою очередь усугубляет дегидратацию и электролитные потери. У части больных на этом фоне могут появиться боли в животе, симулирующие острые хирургические заболевания. Характерно наличие глубокого, шумного дыхания типа Куссмауля, запаха ацетона в выдыхаемом воздухе. На фоне выраженной дегидратации развивается шоковый синдром с клеточной гипоксией и нарушением микроциркуляции. Результатом сложных нарушений метаболизма является расстройство функций жизненно важных органов - - несостоятельность гемодинамики, развитие острая почечная недостаточность(ОПН) и др. Следует отметить, что полной потери сознания может не быть даже при очень тяжелом состоянии больного. Патологический процесс носит прогрессирующий характер, при отсутствии патогенетического лечения дезорганизация метаболизма достигает крайней степени и ведет к гибели больного.
Лабораторные данные: гипергликемия более 300 мг% (18 ммоль/л), глюкозурия, выраженная ацетурия ++++, ацидоз (легкий ацидоз - - рН 7,3-7,2; выраженный ацидоз - - 7,2—7,0; тяжелый ацидоз - - 7,0 и менее, рН :z 6,8 - - несовместимо с жизнью). В периферической крови: гиперлейкоцитоз (13-35)хЮ9/, со сдвигом влево; повышение креатинина (0,2-0,5).
Гиперосмолярная кома. Более тяжелое состояние, чем ДКА, встречается
значительно реже. При гиперосмолярной диабетической коме
нарушение деятельности ЦНС проявляется энцефалопатией, переходящей в кому. Нарушения функции ССС клинически проявляются тахикардией, снижением АД вплоть до развития коллапса, анурией. Часты тромбоэмболические осложнения. Особенностями гиперосмолярной комы являются отсутствие кетоацидоза, умеренная выраженность электролитных расстройств при выраженной гликемии и связанной с этим дегидратации.
Лабораторные данные: гипергликемия (выше 50 ммоль/л), повышение ге-матокритного числа, лейкоцитоз менее выражен, рН крови в норме (7,35), кре-атинин повышенный (идет катаболизм белка); в моче ацетона нет или один +.
Гиперлактацидемическая кома, лактацидоз - - вид расстройства метаболизма, возникающий при патологических состояниях, сопровождающихся гипоксией (СП, дыхательная недостаточность, физические перегрузки и др.), активизирующей анаэробный путь превращения глюкозы, с образованием молочной кислоты. У больных диабетом лактацидоз развивается при лечении бигуанидами лиц, имеющих противопоказания к применению препаратов этой группы (гипоксия любого генеза, алкоголизм, инфекции и др.).
В клинической картине гиперлактацидемической комы имеют место явления ацидоза и интоксикации. Падение рН крови ниже 7,2 может сопровождаться гемодинамическими нарушениями из-за снижения чувствительности рецепторного аппарата миокарда и сосудов к вазопрессорному действию катехоламинов. При лабораторных исследованиях выявляется гипергликемия (часто умеренная), снижение рН крови, повышение концентрации молочной кислоты.
Гипогликемическое состояние и кома возникают при передозировке лекарственных средств (инсулина или сульфаниламидных препаратов) и (или) нарушении режима питания. Предрасполагает к развитию гипогликемии почечная и печеночная недостаточность, мышечная работа, употребление алкоголя. Гипокалиемия, как правило, развивается внезапно. Больной ощущает беспокойство, чувство голода, тремор, потливость, головную боль, слабость. Если помощь своевременно не оказана, то состояние прогрессивно ухудшается, нередко возникают возбуждение, неадекватноость поведения, судороги, потеря сознания. АД повышено, отмечается тахикардия, профузный пот.
Нарушения чувствительности
Все виды чувствительности от кожи, мышц, суставов, сухожилий передаются в ЦНС через три нейрона.
1. Первый – в спиномозговых узлах.
2. Второй – в задних рогах спинного мозга (болевая и температурная) или в тонком и клиновидных ядрах продолговатого мозга (глубокая и тактильная чувствительности). Располагаются в ядрах Голля и Бурдаха.
3. Третий – в талямусе от него аксоны поднимаются к чувствительным зонам коры мозга.
Виды: световая, звуковая, тактильная, вкусовая, температурная, болевая, обонятельная, глубокая (мышечно-суставная).
Более сложные, контролируемые высшими корковыми центрами, чувства локализации, кинетическое, двухмерно-пространственное.
Этиология
Травма, опухоли, воспаление, кровоизлияние, интоксикация.
Феномены нарушения чувствительности:
1. Боль.
2. Гипестезия.
3.Анестезия: аналгезия, термоанестезия, топанестезия, батианестезия.
4. Гиперестезия
5. Парестезия.
6. Дизестезия – извращение (холод как тепло).
7. Гиперпатия – любое раздражение сопровождается неприятным чувством.
Нарушения на любом уровне: рецептор – чувствительный нерв – спинной мозг – ретикулярная формация – талямус – кора, называемая деафферентационным синдромом.
Рецепторный механизм связан с изменениями пороговых характеристик: сила и длительность, количество и плотность, распределения рецепторов. На уровне спинного мозга можно наблюдать, при половинной поперечной перегрузке спинного мозга, синдром Броун-Секара, который характеризуется параличом и выпадением мышечно-суставным чувствительности (глубокой) на стороне поражения, болевой и температурной на противоположной стороне. При полном разрыве спинного мозга наблюдается двусторонняя анестезия.
Поражение талямуса
Поражение характеризуется перекрестным снижением или выпадением всех видов чувствительности (гемианестезия). Кроме того, формируются талямические боли (параксизмальные, жгучие, мучительные) в противоположной половине тела.
ПАТОФИЗИОЛОГИЯ НЕВРОЗОВ
Невроз - хроническое нарушение высшей нервной деятельности, вызванное психоэмоциональным перенапряжением и проявляющееся нарушениями интегральной деятельности мозга - поведения, сна, эмоциональной сферы и сомато-вегетативной деятельности. Это психогенное заболевание, возникающее на фоне особенностей личности и недостаточности психической защиты с формиованием невротического конфликта. Невроз охватывает все сферы деятельности организма, это чрезвычайно универсальное явление.
Актуальность этой проблемы связана с непрерывным ростом числа больных людей - за последние 70 лет частота неврозов возросла в 25 раз!!!
Психогенным фактором во всех случаях являются внешние или внутренние конфликты, действие психотравмирующих обстоятельств или массивное перенапряжение эмоциональной или интеллектуальной сфер психики.
Наряду с экзогенными факторами (трудные задачи, конфликтные ситуации) в развитии патологических состояний ВНД важную роль играют также и эндокринные факторы, среди которых на первый план выступают эндокринновегетативные влияния. Среди эндокринных факторов особое место занимают половые гормоны. Связь нервных расстройств с нарушением половых функций была известна еще в глубокой древности.
Классификация неврозов. Истинные или психогенные, большие неврозы: неврастения, истерия и невроз навязчивых состояний. Невроз - болезь целого организма, что находит отклик в нарушении деятельности различных систем организма. Невроз маскируется под разнообразные соматические болезни: ишемическая болезнь сердца, инфаркт миокарда. Многие больные (до 90%) имеют неврологический компонент и его необходимо обязательно лечить. От неврозов необходимо отличать неврозоподобные состояния, которые при сходной с неврозами клинической картине имеют органические поражения НС, а не психоэмоциональное перенапряжение. Например эндокринные нарушения - гипер- или гипотиреоз влияют на вегетатику, или гипертоническая болезнь, или пептическая язва.
И еще психозы - тоже неврозы, но при неврозе больной критически оценивает себя, знает, что он болен и может преувеличивать свою болезнь, то при психозе больной все отрицает.
Этиология неврозов. Исторически сложились 2 направления в изучении неврозов:
1) физиологическое - исследование ВНД физиологическими приемами (И.П.Павлов) и
2) психофизиологическое изучение - Зигмунд Фрейд.
Современная психиатрия рассматривает невроз как следствие психогенного конфликта. В настоящее время ни в клинике, ни в эксперименте нет еще полной ясности, с чего начинается невроз, какие конкретные церебральные механизмы нарушаются раньше и какие позднее; каково удельное значение отдельных структур в этой патологии. Правда, благодаря блестящим работам И.П.Павлова и его школы еще в двадцатых годах было установлено, что невроз - это срыв ВНД вследствие перенапряжения нервных процессов или их подвижности с нарушением межцентральных корково-подкорковых отношений, отражением которых на периферии являются различные вегетативно-висцеральные расстройства.
Среди причин неврозов выделяют 3 группы - биологические, психологические и социальные. К биологическим относятся наследственность и конституция, беременость и роды, пол, возраст, перенесенные заболевания. К психологическим факторам относят преморбидные особенности личности, психические травмы детского возраста, психотравмирующие ситуации. А родительская семья, особенности сексуального воспитания и семейное положение, образование, профессия, трудовая деятельность рассматриваются в группе социальных факторов. Однако правильнее их рассматривать как факторы предраспологающие, способствующие реализации этиологического фактора, каким и является психическая травма. Опыт изучения неврозов выявляет значение двухэтапной психической травматизации. У подавляющего большинства обследованных больных были "детские психогении" - неполная семья, конфликтные отношения родителей, потеря значимых объектов, нарушение эмоционального контакта с одним или обоими родителями, аморальное поведение родителей. И у части больных на этом фоне возникали характерные для детей формы невротического реагирования;энурез, логоневроз, тикозные гиперкинезы. У других видимые невротические расстройства отсутствовали. Затем возникали актуальные психогении, носившие характер "второго удара". По-видимому, существует определенный фактор, который определяет индивидуальную реакцию на казалось бы однозначные внешнесредовые ситуации.Этим фактором является значимость для индивидуума психогенного воздействия.
Вопросы патогенеза неврозов. На фоне взаимодействия психической травмы и особенностей структуры личности формируется ключевое звено патогенеза - невротический конфликт. Формирование, а в дальнейшем и разрешение конфликта тесно связано с состоянием защитных механизмов личности. Существующие вне сферы сознания установки играют роль в выборе поведения. Наличие подвижных, быстро приспосабливаемых к меняющимся условиям среды установок является фактором, противодействующим возникновению невроза или способствующим успешному разрешению невротического конфликта. Исследование сна показало его защитное психологическое значение. Кроме того, огромную роль играют нейрофизиологические, нейрохимические, психофизиологические и морфологические аспекты. Формирование неврозов связано с типом ВНД.
По Павлову выделяют 4 типа ВНД на основе силы, подвижности и уравновешенности основных нервных процессов (возбуждения, торможения, подвижности).
Что же такое тип ВНД? Это совокупность врожденных и приобретенных свойств НС, определяющих характер взаимодействия организма с окружающей средой, находящий свое отражение во всех функциях организма. При этом удельное значение врожденного и приобретенного в фенотипе может меняться в зависимости от условий взаимодействия организма со средой. В обычных условиях в поведении человека и животного доминируют индивидуальный опыт, привычки, приобретенные навыки. Однако когда организм попадает в необычные - экстремальные - условия, то на первый план в его поведении выступают преимущественно врожденные механизмы нервной деятельности.
Виды неврозов:
Неврастения - бессилие, нервное истощение - самый распространенный невроз, основным проявлением которого является состояние раздражительной слабости, повышенная истощаемость и замедленность восстановления психических процессов.
В начале заболевания (гиперстенической стадии) периодически возникают колебания настроения,общая гиперестезия,повышенная раздражительность. При этом даже незначительные раздражители: громкий разговор, скрип двери и т.п.выводят больного из равновесия - он не может сдержить себя, повышает голос. Больные жалуются на трудность засыпания, вегетодистонии - повышенную потливость, сердцебиения, головные боли. Эти расстройства исчезают при лечении.
Вторая (промежуточная) стадия проявляется более стойким состоянием раздражительной слабости, повышенной эмоциональной возбудимости, несдержанностью, непереносимостью ожидания на фоне повышенной психической истощаемости, ослабления активного внимания, быстрым переходом к чувству усталости, нередко со слезами. Затрудненное засыпание может сопровождаться тревожным ожиданием бессонницы; характерен поверхностный сон с тревожными сновидениями, после которого больной чувствует себя невыспавшимся. Характерны вегетативные расстройства - жалобы на сердце, кишечник как на органическую патологию. Третья стадия (гипостеническая), которой свойственны резкая истощаемость, вялость, адинамия, апатия.
В начальной стадии неврастении преобладает слабость внутренного торможения, во второй - процесс возбуждения начинает ослабевать и становится патологически лабильным с быстрой истощаемостью и в третьей стадии развивается слабость обоих нервных процессов с преобладаением запредельного торможения. По мнению И.П.Павлова, к неврастении склонны лица со слабым типом нервной системы.
Истерия - разновидность психогений, возникающая в связи с психотравмирующей обстановкой у лиц с истерическим складом характера и у здоровых ранее лиц в тяжелых экстремальных условиях. Чаще появляется в молодом возрасте, преимущественно у женщин. Выражается в многочисленных функциональных расстройствах, внешне напоминающих самые различные болезни, за что получила название "хамелеон" ,"великая симулянтка". Особенность больных с истерией - стремление любым путем привлечь к себе внимание окружающих и очень большая внушаемость и самовнушаемость.
Симптоматику истерии условно можно подразделить на двигательные, сенсорные, вегетативные и психические расстройства. Двигательные - выражаются в виде истерических припадков,парезов, мышечных контрактур, различных нарушений походки, заикания. Истерический припадок возникает в чьем-либо присутствии, проявляется падением, обычно неопасным в виде медленного опускания с разнообразными движениями, криками, своеобразными позами с характерной мимикой, но сознание не теряют. Припадок может быть прерван внешним воздействием и нередко переходит в плач, состояние разбитости, усталости и реже в сон.
Истерические сенсорные расстройства могут проявляться снижением чувствительности до полной анестезии на тактильные, температурные, болевые раздражители либо гиперестезиями на те же реагенты.
Вегетативно-висцеральные расстройства очень многообразны; может быть чувство сжатия гортани - комок в горле, ощущение нехватки воздуха (напоминающее бронхиальную астму), ощущуние непроходимости пищевода, задержка мочеиспускания, запоры. Возможны парезы кишечника, напоминающие кишечную непроходимость. Нарушения со стороны сердечно-сосудистой системы, симулирующие стенокардию или инфаркт миокарда. Возможны истерические обмороки и т.п.
Со стороны психики более типичны психогенные амнезии, тотальные или частичные. Истерические галлюцинации - очень яркие, образные, красочные. Возможны бредоподобные фантазии.
Симптоматику истерии И.П.Павлов объяснял характерным превалированием подкорковой деятельности над корковой и первой сигнальной системы над второй.
Навязчивые состояния - мысли, сомнения, действия, страхи, движения, возникающие независимо и вопреки желанию больного, притом непреодолимо. Больные относятся к ним критически, понимают их бессмысленность и болезненный характер, но освободиться не могут.
Навязчивые страхи (фобии) встречаются очень часто и в самой разнообразной форме. Наиболее распространенные из них следующие:
Агорафобия - страх открытого пространства.
Акрофобия - страх высоты.
Дисморфофобия - страх уродства.
Клаустрофобия - страх замкнутых пространств, закрытых помещений.
Нозофобия - страх заболеть какой-нибудь тяжелой болезнью. Сюда относится акарофобия (страх чесотки), бактериофобия, канцерофобия.
Танатофобия - страх смерти, тафефобия - страх быть заживо погребенным. В группе навязчивых страхов могут быть выделены особо навязчивые опасения - невозможности совершения какого-либо обычного жизненного или профессионального акта. Певица боится, что не споет хорошо известную арию и отказывается от выступления. При навязчивых воспоминаниях в сознании больного мучительно вновь и вновь возникает образное воспоминание о каком-то неприятном, порочащем его событии. В н-х - синдромы:
1. астенический - слабость нервной деятельности;
2. истерический – эмоциональная несдержаность;
3. депрессивный - угнетение, боязнь;
4. фобический - страх, опасения;
5. ипохондрический - жалобы на деятельность внутренних органов.
Экспериментальные модели неврозов (с 1921 г.И.П.Павлов):
1) У животных вырабатывают возбудительные и тормозные пищевые рефлексы (+) в круге - пища, эллипс с (-) - дифференцировочный образ без пищи, формируется активное внутренне торможение и при соотношении осей эллипса 7:8 животное не различает его от круга - возникает бурная реакция - лай, беспокойство и на несколько месяцев нарушаются условные рефлексы в связи со срывом ВНД - невроз.
2) Перенапряжение силы нервных процессов (возбуждения) при действии сверхсильных раздражителей, большого числа раздражителей.
3) Перенапряжение активного тормозного процесса при удлинении действия тормозного раздражителя с 30 сек до 10 мин.
4) Перенапряжение подвижности нервных процессов - "сшибка" - столкновение разнородных рефлексов (+) и (-). Звонок №1 (-) без еды и через 5 минут звонок №2 (+) - еда. Если между звонками пауза 5 мин - все нормально, но если звонки следуют друг за другом - сталкиваются процессы возбуждения и торможения - основной прием получения неврозов.
Позднее Павлов разработал 3 модели неврозов,адекватных человеческим:
5) Столкновение биологически противоположной деятельности "сшибка" вырабатывают условный пищевой рефлекс на раздражение кожи слабым электрическим током и затем увеличивают силу тока - боль и пища.
6) Переделка динамического стереотипа условнорефлекторной деятельности - группа раздражителей различного знака друг за другом следуют в одинаковом порядке и с одинаковыми интервалами в 5 мин (М-метроном):
М 120 (+),
М 60 (-),
свет (+),
звук №1 (+),
звук №2 (-),
будильник (+),
но при смене порядка подачи раздражителя или изменении времени подачи его легко возникает невроз. Деятельность человека всегда стереотипна, это проще и у большинства людей переделка жизненного стереотипа вызывает невроз.
7) Информационные неврозы - от обилия жизненно важной информации при недостатке времени на ее полноценную переработку: вырабатывают 4 сложных стереотипа условнорефлекторной деятельности у животных в камере 1 2 и по окончании соответствующего последнего сигнала животное 4 3 получает пищу в определенной кормушке из 4-х. Если промежутки времени между стереотипами большие - несколько часов - животное бежит точно к нужной кормушке, но при сближении времени стереотипов происходит срыв, ошибки, взрыв эмоций. Человек получает очень много жизненной информации и не успевает переработать ее → невроз в связи со срывом ВНД.
8) Даже фиксация животных в течение полугода в станке вызывала нарушения условнорефлекторной деятельности - ведь изымалось движение.
Оказалось, что возникновение неврозов зависит от типа ВНД. Для слабого типа нервной деятельности любое перенапряжение вызывает невроз. У безудержного нужно перенапрягать тормозные процессы (круг/эллипс), у инертного – нужно перенапрягать подвижность (сшибка), у уравновешенного получить невроз гораздо сложнее. И.П.Павлов считал неврозы следствием перенапряжения и срыва ВНД.
Так молодая семья живет вместе со свекровью и длительное время молодая жена не реагирует на замечания свекрови и все нормально. Но через несколько лет по пустяку взрыв эмоций - истерический невроз из-за многолетнего активного центрального торможения (опыт физиологического понимания).
И это особенно важно для стариков - переезд на новую квартиру, выход на пенсию - ломка стереотипа.
Патогенез экспериментальных неврозов. Клиническая картина почти всех форм неврозов включает в себя, как правило, нарушение сна, вегетативно-висцеральные, преимущественно сердечно-сосудистые, расстройства. Это, естественно, направляет внимание исследователей, пытающихся найти локальный адрес невротических нарушений, к структурам лимбического или так называемого висцерального мозга, и прежде всего к эмоциогенным отделам гиппокампа, corpus amygdaloideum , гипоталамуса. В последнее время все чаще встречаются указания на важную роль в патогенезе неврозов структур лимбико-ретикулярного комплекса, с которым связана основная симптоматика болезни. Кроме того, общепризнано, что для развития невроза, помимо стресса, должна быть еще и генетически или прижизненно обусловленная предрасположенность. В связи с этим научно-техническую революцию с ее "информационными перегрузками" и пр.следует рассматривать не как причину, а как условия, астенизирующие нервную систему и тем самым предрасполагающие к развитию неврозов.
Нарушения условнорефлекторной деятельности после невротизирующих воздействий были во всех случаях у всех животных, но выражадись они поразному: в виде увеличения латентных периодов и нарушения силовых отношений рефлексов с развитием фазовых состояний (уравнительная, парадоксальная, ультрапарадоксальная), снижения или выпадения условных рефлексов и т.п. Четко выявлялась завивисимость характера нарушений условнорефлекторной деятельности от типологических особенностей нервной системы. Нарушения эти были длительными и носили, особенно в начале болезни, волнообразный характер:периодическое улучшение без всякой видимой причины вновь сменялось ухудшением. Эти волнообразные изменения состояния ВНД ученые предлагают рассматривать не как проявление начинающейся болезни, а скорее как мобилизацию защитных сил организма. Изменения со стороны вегетативных функций наблюдались у всех животных и проявлялись по-разному у представителей разных типов ВНД.
Со стороны эндокринной системы было показано, что введение гидрокортизона в среднетерапевтических дозах у собак сильного типа НС повышает условные рефлексы, улучшает дифференцировку, тогда как у собак слабого типа эти дозы ухудшают условнорефлекторную деятельность, снижая условные и безусловные рефлексы. Хроническое применение кортизона (как и АКТГ) приводит к длительным нарушениям ВНД у животных и после прекращения введения препаратов. Эти гормоны рассматриваются как непременные компоненты стрессовых реакций, они "запускаются" под влиянием адреналина, выделяемого при любых стрессовых ситуациях.Как правило, введение малых доз гормонов: тиреотропного, АКТГ, кортизона, половых гормонов, адреналина - оказывает стимулирующее действие на ВНД, а высокие дозы гормонов угнетают ее, нарушая условнорефлекторную деятельность.
На ЭКГ у собак при экспериментальном неврозе наряду с увеличением ЧСС регистрировалась экстрасистолия, сглаженность или даже выпадение зубца P, увеличение или двухфазность зубца T, увеличение зубца R.
На ЭЭГ усиление тета - и альфа-частот во всех структурах.
Вообще факторами, способствующими повышению мозгового кровотока при эмоциональном стрессе и усилению вегетативных реакций, могут быть катехоламины, выделяемые катехоламинергическими системами и надпочечниками. Известно, что с повышением артериального давления гематоэнцефалический барьер становится проницаемым для катехоламинов, которые увеличивают скорость метаболических процессов и мозговой ткани и повышают локальный мозговой кровоток (ЛМК).При невротических нарушениях, вызванных длительными стрессовыми воздействиями, происходит истощение катехоламиновых систем, что может привести к снижению интенсивности метаболических процессов и снижению ЛМК. Отмечены нарушения всех фаз сна - укорочение продолжительности глубоких фаз сна, увеличено число пробуждений - его дефектность и функциональная неполноценность. Были выявлены нейромедиаторные нарушения, были сосудистые и глионейрональные нарушения, указывающие на развитие в ЦНС гипоксии. Было выявлено снижение скорости локального кровотока в 2-3 раза.
Психопатологичесое направление (основоположник Зигмунд Фрейд) - в основе неврозов лежит нарушение бессознательной психической деятельности человека - инстинкты: любовь и агрессия. Фрейд выделил 3 уровня: бессознательный, подсознательный и уровень сознания. Фрейд считал источником неврозов подавление бессознательной деятельности, т.к. у человека она постоянно сдерживается уровнем сознания. Воспитание людей - это постоянное ограничение инстинкта и это (по Фрейду) ведет к неврозам. Инстинкт не может исчезнуть, и когда его подавляют - то он проявится искаженным - в виде невроза (по Павлову - "сшибка"). Фрейд предложил способ писхоанализа:
1) анализ поведения;
2) анализ ошибочных действий человека;
3) свободное изложение мыслей, которые приходят в голову,когда человека о чем-либо спрашивают - метод свободных ассоциаций. Больного нужно очистить от переживаний, навязчивых мыслей.
Итак, причина невроза - хронический психоэмоциональный стресс, связанный с перенапряжением ВНД - смыкание физиологического и психоэмоционального направления.
Схема патогенеза неврозов: психоэмоциональный стресс → стимуляция мозговой деятельности; стрессорные реакции → нарушение интегративной деятельности (дезинтеграция нервной деятельности, нарушения поведения и сна) → нарушения вегетативной нервной деятельности, нейромедиаторной активности, эндокринной системы (симпатоадреналовые сдвиги, увеличение выработки дофамина, ваготонии, инсуллярные сдвиги) → нарушения метаболизма микроструктур и микроциркуляции → нарушения деятельности внутренних органов и соматической сферы. Формируется порочный круг – гипоксия мозга стимулирует психоэмоциональный стресс и стимулирует деятельность мозга.
У детей неврозы характеризуются малой очерченностью, стертостью, большой изменчивостью клинических признаков. Отсутствуют классические формы, кроме истерических и фобических; преобладает двигательная расторможенность. Отсутствуют отчетливые жалобы со стороны ребенка и обилие их от окружающих. Есть основной симптом или синдром, определяющий особенности болезни (так называемый моносимптоматический невроз); изменение поведения и снижение успеваемости.
Неврозы у детей характеризуются наличием отчетливых предрасполагающих факторов, способствующих возникновению невроза, благоприятным течением и прогнозом. Они имеют следующие особенности: чем меньше возраст ребенка, тем меньшая дифференциация невроза, тем чаще его картина представлена преходящими невротическими реакциями. С возрастом картина невроза становится все более типичной, клинически более очерченной. Эмоциональные переживания ребенка фиксируются на деятельности внутренних оганов и систем. Детям свойственна также большая фиксация на конфликтной ситуации, что легко приводит к возникновению страха, например: страха темноты, одиночества, расстройства аппетита.
В преклонном возрасте имеет место та же картина невроза, что и в детстве, однако с противоположной динамикой.
Профилактика неврозов:
1) прекращение хронического действия стрессора - все перемелется;
2) наличие высоких целей в жизни и реальных возможностей для их достижения;
3) создание философии жизни - все умрем - радуйся, пока живешь;
4) сделай так, чтобы ближний полюбил тебя как себя самого.
ЛИТЕРАТУРА
1) Патологическая физиология. Адо А.Д., Л.М. Ишимова. М.,1973
2) Патологическая физиология. Адо А.Д, Новицкого В.В Томск,1994
3) Патологическая физиология. П.Ф. Ливицкого. М.,1995
4) Патологическая физиология. Н.Н. Зайко и др. Киев,1996
5) Патологическая физиология Н.Н. Зайко., Ю.Б. Быця. Киев,2002
6) Патологическая физиология А.И. Воложин. М., 1998.
7) Основа общей патологии. А.Ш. Зайчик. С-Пб., 1999.
8) Патологическая физиология. Адо А.Д. и др. М., 2000.
9) Патологическая физиология. Адо А.Д., М.А. Адо, В.И. Пыцкий,
Г.В. Порядин, Ю.А. Владимиров. М.,2001.
10) Патологическая физиология. В.А. Фролов и др. М.,2002
11) Патологическая физиология. В.А. Фролов, Г.А. Дроздова, Т.А.
Казанская, Д.П. Билибин, Б.А. Демуров. М.,1997.
12) Практикум патологическая физиология. В.Ю. Шанина. С-Пб,2002
13) Механизмы развития болезней и синдромов. А.Ш. Зайчик. С-Пб,2002
14) Патологическая физиология. (Том 1,2). П.Ф. Литвицкий. М.,2002-2003.
15) Патологическая физиология в рисунках, таблицах и схемах.
В.А.Фрололва. М.,2003.
16) Введение в экспериментальную патологию (практикум). А.Ш. Зайчик и
др. С-Пб.,2003.
17) Патологическая физиология Н.Н. Зайко., Ю.Б. Быця. М.,2004
БАШКИРСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
ГИЗАТУЛЛИН А.Г.
КРАТКАЯ ПАТОФИЗИОЛОГИЯ
ОРГАНОВ И СИСТЕМ
ЛЕКЦИИ
(Частная патофизиология)
Г .
ОГЛАВЛЕНИЕ
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ ДЕЯТЕЛЬНОСТИ.. 2
Дата: 2019-03-05, просмотров: 444.