Глава 10
ЭЛЕКТРОЛИЗ
Электролиз является одним из основных методов получения редких металлов, сплавов и рафинировки чернового металла.
Электролизом называется разложение электролитов постоянным электрическим током, которое сопровождается образованием новых веществ. На электродах происходят реакции окисления-восстановления: анионы на аноде отдают электроны и окисляются, а катионы восстанавливаются на катоде. Если анод растворим в электролите под действием тока, то чаще всего анионы на нем не разряжаются, а электронейтральность раствора (или расплава) поддерживается образованием катионов из материала анода. Одно из преимуществ электролиза перед химическим восстановлением заключается в том, что при этом продукты восстановления не загрязняются остатками металла-восстановителя и примесями, первоначально присутствующими в нем. Кроме того, при электролизе возможна очистка от многих примесей исходного сырья. Изменяя условия электролиза, можно получать катодный осадок с некоторыми заданными физическими свойствами (крупностью кристаллической структуры и т.п.). В промышленных масштабах осуществляют электролиз как водных растворов, так и расплавов. Однако для получения редких металлов электролиз водных растворов используют редко.
В табл. 34 приведены значения нормальных (стандартных) электродных окислительно-восстановительных потенциалов в водных растворах.
Таблица 34
Стандартные электродные окислительно-восстановительные потенциалы при 25° С в водных растворах
| Система | Eo, в | Система | Eo, в |
| Li = Li++ e- | -3,045 | Тm = Тm3+ + 3e- | -2,278 |
| К = К++ e- | -3,925 | Lu = Lu3+ + 3e- | -2,255 |
| Rb = Rb+ + e- | -2,924 | Sc = Sc3+ + 3e- | -2,077 |
| Na = Na+ + e- | -2,714 | Th = Th4+ + 4e- | -1,0899 |
| La = La3+ + 3e- | -2,522 | Be = Be2+ + 2e- | -1,847 |
| Pr = Pr3+ + 3e- | -2,462 | Hf = Hf4+ + 4e- | -1,700 |
| Nd = Nd3+ + 3e- | -2,431 | Al = Al3+ + 3e- | -1,663 |
| Sm = Sm3+ + 3e- | -3,121 | Ti = Ti2+ + 2e- | -1,630 |
| Eu = Еи2+ + 2e-- | -3,395 | Zr = Zr4+ + 4e- | -1,539 |
| Gd = Gd3+ + Зe - | -2,397 | V = V2+ + 2e- | -1,175 |
| Tb = Тb3+ + 3e- | -2,391 | Nb = Nb3+ + 3e- | -1,1 |
| Mg = Mg2+ + 2e- | -2,363 | 2Ta +5H2O = Ta2O5 + 10H+ + 10e- | -0,750 |
| Y = Y3+ + 3e- | -2,372 | Ni = Ni2+ + 2e- | -0,250 |
| Dy = Dy3++ 3e- | -2,353 | H2 = 2H2+ + 2e- | 0,000 |
| Ho = Ho3+ + 3e- | -2,319 | Cu = Cu2+ + 2e- | +0.337 |
| Ег = Ег3+ + Зe- | -2,296 |
Получение из водных растворов электроотрицательных металлов, расположенных выше водорода, невозможно, поскольку при пропускании тока через раствор выделяется водород. Лишь в некоторых случаях, когда перенапряжение водорода на поверхности выделяемого металла достаточно велико, электролиз возможен (например, выделение никеля из водных растворов). Возможен также электролиз на жидком ртутном катоде с получением амальгамы, так как перенапряжение водорода на ртути значительно.
Как видно из табл. 34, все редкие металлы, рассматриваемые в настоящем учебном пособии, электроотрицательнее водорода, вследствие чего для них приобретает особое значение процесс электролиза безводных и расплавленных сред.
При электролизе расплавов целесообразно получать металл в расплавленном состоянии, поскольку в таком виде его легче выводить из ванны (непрерывный процесс); катодный продукт после охлаждения представляет собой компактный слиток (уменьшение включений электролита). Однако большинство редких металлов (см. стр. 291) относится к числу тугоплавких. Поскольку отсутствуют доступные соли, не улетучивающиеся или не разлагающиеся при температуре выше 1500° С, тугоплавкие металлы обычно получают при температуре, более низкой, чем точка их плавления. Повышение температуры до расплав-ления металла часто связано с непреодолимыми аппаратурными затруднениями.
Металлы при электролизе в условиях tэл < tпл осаждаются на катоде в виде кристаллической губчатой массы, растущей по направлению к аноду и содержащей большое количество электролита. Опыты по получению в таких условиях гладких катод-ны.х осадков (по аналогии, например, с электролизом водных растворов меди) не дали положительных результатов. Переработка катодного продукта включает обязательную стадию отделения электролита, который часто содержит нерастворимые в воде соли. Сложность отделения электролита приводит к потерям мелкодисперсной фракции металла, и извлечение металла снижается. Иногда этих затруднений можно избежать при электролизе на жидком катоде — металле с невысокой температурой плавления. При этом образуется жидкий сплав, который либо можно использовать как таковой, либо после отгонки вспомогательного металла получить губку основного металла.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
При погружении металла в раствор электролита (или расплав) катионы могут переходить из кристаллической решетки металла в раствор (поверхность заряжается отрицательно) или из раствора в металл (поверхность приобретает положительный заряд). Возникает разность потенциалов между металлом и раствором (расплавом). При переходе катионов в раствор возникающий отрицательный заряд поверхности металла препятствует дальнейшему растворению. Поэтому величина скачка (разность потенциалов) между металлом и раствором постоянна в данных условиях.
Абсолютное значение электродного потенциала измерить невозможно, поэтому измеряют всегда разность электродных потенциалов — относительный электродный потенциал.
Если электродная реакция проходит в равновесных обратимых условиях (при токе, стремящемся к нулю), скачок потенциала между электродом и электролитом называют равновесным потенциалом. Если к погруженному в раствор металлу приложить напряжение, на бесконечно малую величину превышающее равновесный потенциал, но обратного знака, процесс, определяющий равновесный потенциал, пойдет в обратную сторону. Если первоначально металл растворялся, то произойдет выделение его на электроде - электролиз. Однако продолжительный электролиз в таких условиях осуществить не удается, так как происходящее нарушение электронейтральности раствора (выведение положительно заряженных ионов) мгновенно создаст противо-э. д. с., процесс прекратится. Для осуществления продолжительного электролиза необходимо производить одновременную разрядку отрицательных ионов раствора на втором электроде (аноде) или восполнение убыли положительных ионов. за счет растворения анода. Поскольку анод, погруженный в раствор (расплав), также обладает определенным потенциалом, то для осуществления электролиза в равновесных условиях необходимо приложить внешнее напряжение, равное сумме равновесных потенциалов анода и катода плюс бесконечно малая величина. Сумма равновесных потенциалов анода и катода называется напряжением разложения.
Равновесное значение потенциала определяется уравнением Нернста:
для катиона Ек = Еко + RTlnaMen+ / nF ; (73)
для аниона Ек = Еко - RTlnaMen- / nF ; (74)
где Е° - стандартный электродный потенциал (при активности ионов, равной 1); n - валентность; а - активность; R - газовая постоянная [8,31 дж/(моль.град)]; Т - абсолютная температура; F - число Фарадея (96520 к/г-экв).
Относительный стандартный электродный потенциал обычно приводится в таблицах для водных растворов по отношению к водородному электроду.
В случае контакта с раствором амальгамы или сплава
Е = Ео +RTln [aMen+/aMe] / nF ; (75)
где aMe - активность металла в амальгаме или сплаве; aMen+ - активность ионов металла в растворе.
Посторонние ионы (примеси других металлов, а также ионы, вводимые для создания определенной среды), даже если они электроотрицательнее основного иона и не разряжаются на катоде, в некоторых случаях существенно влияют на течение электролиза. Они влияют на коэффициенты активности вследствие образования, например, комплексных соединений, участия в процессах гидратации и дегидратации. Нейтральные ионы могут внедряться в двойной электрический слой.
Ионы металлов, более электроотрицательные, чем ион водорода, могут быть выделены из водных растворов на электроде, обеспечивающем большое перенапряжение для выделения водорода. Кроме того, можно сдвигать потенциал выделения основного металла в электроположительную область, увеличивая его активность в электролите. Регулируя (увеличивая) рН раствора, можно сдвинуть потенциал выделения водорода в электроотрицательную область.
Совместное осаждение ионов основного металла и ионов-примесей приводит к загрязнению катодного осадка. Понижение концентрации ионов-примесей в электролите резко сдвигает потенциал их выделения в электроотрицательную область, поскольку активность ионов-примесей <<1. Понижение концентрации приводит также к увеличению концентрационной поляризации. Поэтому очистка исходной соли имеет большое значение для получения осаждаемого металла высокой степени чистоты.
Напряжение разложения соли (т. е. разность обратимых потенциалов катода и анода) можно рассчитать из термохимических данных для соответствующей реакции. Например, если при электролизе происходит разложение соли МеХ3 с выделением Me и Х2, то напряжение разложения рассчитывают из термохимических данных для реакции Me+3/2 Х2 = МеХ3. Расчет сводится к вычислению энергии Гиббса DСT° реакции. При этом
ET = - DGoT / nF (76)
где n - валентность; F—число Фарадея, равное 96520 к/г-экв; DGoT - энергия Гиббса, дж/моль; Е - напряжение разложения, в.
Для приближенной оценки зависимости потенциала разложения от температуры можно пользоваться выражением
dET/dT = DSoT / nF (77)
считая энтропию DSoT не зависящей от температуры (в интервалах, где отсутствуют фазовые переходы).
Если электролизу подвергается смесь солей и происходит разложение лишь одной из них (разрядка наиболее электроположительного металла смеси), при вычислении DGoT реакции необходимо учитывать активность этой соли:
DGT = DGoT + RT In a . (78)
(если за стандартное состояние принимается расплав индивидуальной соли).
Пример расчета теоретического напряжения разложения. Рассчитать значение теоретического напряжения разложения глиноземно-криолитового расплава, если изменение энтальпии DН при 950° С для реакции 2А1+1,5О2 ® А12О3 равно -392,64 ккал, а изменение DS = -83,6 ккал/ (моль .°К)
Решение:
DG == DH -TDS = -392,64 + (273+950) . 83,6/1000 = -290,3 ккал;
-DG/nFT = Еразл = 290,3/6.23,06 = 2,1 в,
где FT =F/1000 . 4,18 = 23,06 (переход от джоулей к килокалориям).
Равновесный потенциал электрода можно определить непосредственно из эксперимента, используя обычную потенциометрическую схему, однако наличие побочных и вторичных процессов в электродной системе сильно маскирует истинное значение величины Е (к маскирующим процессам относятся пассивация электродов, образование соединений с выделяющимися продуктами и т. д.). В электрохимии водных растворов за нулевой электрод сравнения при определении Еравн исследуемого электрода принимают стандартный водородный. Кроме него в водных растворах употребляют еще несколько электродов сравнения с точно известным потенциалом - каломельный, хингидроновый, хлорсеребряный и др.
В электрохимии расплавленных солей такого универсального электрода сравнения нет. Обычно в качестве стандартного используют электрод типа Ме/МеХ, где Me - свинец, серебро, кадмий, Х - галоген. Основная трудность в этом случае при измерении Еравн в расплавах связана с необходимостью учета скачка потенциала на границе раздела двух солей - соли, в которую погружен изучаемый электрод, и расплава соли электрода сравнения.
Наряду с металлическими электродами применяют газовые электроды, обратимые к анионам. Конструктивно такие электроды представляют собой трубку из чистого графита, через которую пропускают хлор (в случае, если изучается электролиз хлоридов) или водород. Стационарный потенциал такого электрода устанавливается довольно медленно.
При установлении ряда значений напряжения выделения металлов в расплавленных солях возникают и другие трудности. Прежде всего это отсутствие стандартного состояния сравнения для разных солей: не ясно, следует сравнивать электроды при температуре плавления солей или при одинаковой температуре. Большие трудности связаны с выбором нулевого электрода.
В табл. 35 приведены электродные потенциалы некоторых металлов в их расплавленных солях.
Таблица 35
Электродные потенциалы металлов, в, в их расплавленных солях при 700° С, нулевой электрод – водородный
| электрод | анион | ||
| Cl- | Br- | I- | |
| Li/Li+ | -2,36 | -2,40 | -2,41 |
| Na/Na+ | -2,37 | -2,35 | -2,27 |
| К/К+ | -2,51 | -2,53 | -2,41 |
| Ag/Ag+ | +0,18 | -0,10 | -0,43. |
| Ве/Ве2+ | -0,90 | - | - |
| Mg/Mg2+ | -1,59 | -1,58 | -1,47 |
| Cd/Cd2+ | -0,26 | -0,46 | -0,65 |
| А1/А13+ | 0,59 | -0,57 | -0,55 |
| Bi/Bi3+ | +0,38 | +0,19 | -0,13 |
| Sb/Sb3+ | +0,53 | +0,21 | +0,03 |
Вопрос об электродных потенциалах анионов в расплавленных солях разработан слабо. Обычно сравнивают значения напряжения разложения солей с одинаковым катионом для определения положения аниона в ряду напряжений. Проще провести определение напряжения разложения соли, которое равно сумме равновесных потенциалов анода и катода. Для этого снимают зависимость тока, протекающего через ячейку электролиза, от приложенного напряжения. Прямолинейный участок зависимости I от U экстраполируется на нулевую силу тока. Соответствующее напряжение принимается равным потенциалу разложения соли в данных условиях.
В табл. 36 представлены значения напряжения разложения ряда солей.
Таблица 36
Напряжение разложения ЕТ, в, при температуре плавления некоторых галогенидов (определенное методом изучения зависимости тока от напряжения)
| катион | анион | |||
| F- | Cl- | Br- | I- | |
| Li+ | 2,55 | 3,53 | 3,22 | 2,87 |
| Na+ | 2,78 | 3,25 | 2,93 | 2,49 |
| К+' | 2,84 | 3,40 | 3,11 | 2,65 |
| Rb+ | - | 3,60 | 2,76 | 2,35 |
| Cs+ | - | 3,77 | - | 2,50 |
| Be2+ | - | 2,08 | - | - |
| Mg2+ | 2,26 | 2,59 | 2,21 | 1,75 |
| Ca2+ | 2,46 . | 2,28 | 2,82 | 2,50 |
| Sr2+ | 2,51 | 3,41 | 3,08 | 2,68 |
| Ba2+ | 2,82 | 3,44 | 3,05 | 2,36 |
| Cd2+ | - | 1,36 | 1,18 | 1,00 |
| Sn2+ | - | 1,34 | 1,09 | 0,96 |
| Pb2+ | - | 1,25 | 1,10 | 0,79 |
Разбавление индивидуальной соли другой солью с общим анионом приводит к повышению напряжения разложения, поскольку потенциал катода становится электроотрицательнее, а потенциал анода остается практически постоянным. Сдвиг потенциала катода не всегда в этом случае соответствует величине, рассчитанной по закону Нернста, так как активности ионов, как правило, не равны концентрациям.
Обычно положение металла в электрохимическом ряду сохраняется независимо от способа составления ряда и характера аниона. Наиболее электроотрицательными металлами в расплавах (как и в водных растворах) остаются щелочные металлы, затем идут щелочноземельные, элементы третьей группы и тяжелые металлы.
Однако ряд напряжений в расплавленных солях несколько меняется в зависимости от температуры, поскольку, температурные коэффициенты э. д. с. химических цепей различаются между собой. Кроме того, при переходе от одного типа анионов к другому в некоторых случаях изменяется положение металла в ряду. Так, литий в хлоридах, бромидах и иодидах более отрицателен, чем натрий, но во фторидах он положительнее натрия. Особенно большие изменения в порядке электроотрицательности металлов при изменении характера аниона характерны для тяжелых металлов (что связано со сложностью конфигурации электронных оболочек и взаимной поляризуемостью ионов).
Таким образом, электролиз расплава теоретически возможен, если к электродам приложено напряжение, лишь на бесконечно малую величину превышающее напряжение разложения соли. Однако в этом случае ток, протекающий через ячейку, также бесконечно мал, а время электролиза бесконечно велико. Для повышения скорости электролиза подается напряжение, заметно превышающее напряжение разложения. Ток при этом имеет какую-то конечную величину. При прохождении тока через электрохимическую систему происходит сдвиг потенциалов электродов от их равновесного значения: катода - в сторону более отрицательных значений, анода — в сторону более положительных значений - электрохимическая поляризация. По современным представлениям, поляризация - следствие замедленности одного или нескольких процессов, происходящих на электроде при прохождении тока. Различают концентрационную, собственно электрохимическую и химическую поляризацию. Часть общего сдвига потенциала электрода, обусловленную электрохимической поляризацией, часто называют перенапряжением.
Концентрационная поляризация обусловлена изменением концентрации потенциалообразующих веществ в приэлектродном слое. Например, разряд ионов.металла при электролизе понижает их концентрацию в слое расплава, непосредственно примыкающем к поверхности электрода. Величину концентрационной поляризации DEк можно вычислить по уравнению Нернста для концентрационных цепей:
DEк = RT/nF . lnC1/Co (79)
где С1 - концентрация потенциалообразующих ионов в приэлектродном слое; Со - концентрация в толще электролита.
При плотности тока iк на единице площади катода разряжается iк/nF г-ионов катионов, т. е. концентрация катионов в прикатодном пространстве убывает со скоростью iк/nF. Убыль катионов возмещается их переносом током. Поскольку число переноса катиона меньше единицы, поступление катионов в результате переноса током меньше убыли и, следовательно, концентрация катионов в прикатодном слое меньше, чем в толще электролита. Увеличение плотности тока приводит к такому состоянию, когда все подходящие катионы разряжаются и их концентрация у катода равна нулю. В этом случае увеличение приложенного напряжения не приведет к росту плотности тока, так как диффузия в создавшихся условиях максимальна и не в состоянии далее увеличить подвод катионов к электроду.
Постоянная и не зависимая от потенциала плотность тока называется предельной плотностью тока и рассчитывается по уравнению
iпр = DnFC / d(1-nк ) = KдС (81)
где D - коэффициент диффузии; n - валентность; F - число Фарадея; С - концентрация в объеме; d - толщина диффузного слоя; nк - число переноса катиона; Кд - константа скорости диффузии. Кд увеличивается с повышением температуры и скорости перемешивания электролита (в результате уменьшения d).
Концентрационная поляризация связана с плотностью тока уравнением
DEк = RT/nF ln (1- iк/iпр) (81)
Достижение предельного тока для данной примеси способствует очистке катодного осадка, так как дальнейшее повышение приложенного напряжения повышает скорость выделения основного металла при постоянной скорости выделения примеси.
Перемешивание электролита, повышение температуры и прочие факторы, облегчающие подачу вещества к электроду, повышают iпр и снижают концентрационную поляризацию.
Собственно электрохимической поляризацией называется смещение потенциала электрода, обусловленное только замедленностью протекания самого электрохимического процесса. Замедленность связана с тем, что электрохимическая реакция, как и всякая другая химическая реакция, требует определенной энергии активации. Наиболее высокие значения электрохимической поляризации наблюдаются при выделении газов. Возникновение перенапряжения при выделении водорода обычно связывают с замедленностью какой-либо одной или нескольких стадий этого процесса: 1) разряд иона водорода: Н+ + МеН + е- ® МеН (Me - металл, МеН - атом водорода, хемосорбированный на металле); 2) рекомбинация адсорбированных атомов: 2МеН ® Н2 + 2Me; 3) электрохимическая десорбция: Н+ + МеН + e- ® H2 + Me.
При малой плотности тока перенапряжение водорода прямо пропорционально плотности тока:
DЕ = wi
(w - константа). При большой плотности тока зависимость перенапряжения от плотности тока определяется уравнением
DЕ = a + b lg i. (82)
Повышение температуры снижает перенапряжение. Энергия активации электрохимической реакции, связанной с переходом заряда через границу электрод - раствор, изменяется с изменением потенциала электрода. Поэтому, смещая потенциал в нужную сторону (в отрицательную для катода и положительную для анода), можно снизить энергию активации электрохимического процесса и тем самым повысить его скорость до значений, обеспечивающих прохождение тока необходимой плотности.
Иногда из общего сдвига потенциала выделяют химическую поляризацию, которая определяется процессами, изменяющими химический состав поверхности электрода: покрытие поверхности анода пленкой труднорастворимых окисей, образование сплавов и интерметаллических соединений на катоде и т. п. Если концентрационную поляризацию легко определить, сравнивая, например, величины сдвига потенциала при различной скорости перемешивания электролита, то разделение электрохимической и химической поляризации часто невозможно, так как одно и то же явление может быть причиной возникновения или увеличения поляризации обоих видов. Так, образование пленок окислов приводит как к закрытию части поверхности электрода, так и к повышению энергии активации отдельных стадий процесса.
Поляризация возрастает с увеличением плотности тока, уменьшается с ростом температуры и зависит от природы электрохимической реакции, материала электрода, состава электролита и других факторов. При электролитическом осаждении металлов на катоде, как правило, большая поляризация не наблюдается.
В процессе электролитического рафинирования металла растворяется черновой анод. При этом поляризация обусловливается возрастанием концентрации ионов металла в прианодном пространстве. Пределом возрастания концентрации является насыщение и последующее выпадение осадка соли на поверхности анода, что приводит к резкому возрастанию сопротивления и падению плотности тока (солевая пассивация анода). Примеси, входящие в черновой анод, могут образовывать самостоятельные фазы. При этом потенциал анода определяется наиболее электроотрицательным компонентом смеси, более положительные компоненты не растворяются.
Частицы нерастворившихся компонентов по мере растворения электроотрицательного компонента, потеряв связь с анодом, падают на дно ванны в виде шлама.
Если электроотрицательный металл составляет меньшую часть массы анода, растворение его идет до тех пор, пока он присутствует на поверхности электрода. Затем (если поддерживать постоянную плотность тока) потенциал электрода возрастает, и начинает растворяться более благородный металл. Более сложная картина наблюдается в случае образования компонентами анода твердых растворов и химических соединений.
Электрохимическое выделение газов на аноде связано с поляризацией, причини которой установлены не во всех случаях. При выделении хлора на угольном электроде наблюдается поляризация, достигающая 0,1 в. Предполагают, что она связана с процессом адсорбции молекулярного хлора. Возможно, перенапряжение связано с замедленностью процесса образования молекул хлора из атомов.
Если выделяющийся металл взаимодействует с материалом катода, происходит деполяризация катодного процесса — сдвиг потенциала катода, в электроположительную сторону. Особенно сильна деполяризация при выделении одного жидкого металла на другом с образованием сплава. Свободная энергия реакции разряда ионов уменьшается на величину энергии взаимодействия металлов, что приводит к значительной деполяризации. Деполяризатором анодного процесса обычно служит металл, растворенный в электролите и взаимодействующий с анодными продуктами.
При электролизе возникновение поляризации нежелательно, так как приводит к дополнительному расходу электроэнергии из-за возрастания напряжения на электролитических ваннах.
Осаждение металлов на катоде в расплавленном состоянии происходит чаще всего при равновесном потенциале. Использование сложных электролитов способтсвует появлению поляризации. Осаждение твердых металлов сопровождается химической поляризацией, а также иногда поляризацией, связанной с замедленностью образования и роста кристаллов. Условия электролиза влияют на характер катодного осадка: при низкой плотности тока на поверхности кристаллов может образоваться окисная пленка - пассивация катода, увеличение поляризации. Таким образом, для практического осуществления процесса необходимо на электролизер подать напряжение, величина которого складывается из нескольких составляющих:
Uпракт = Еa – Ек + DЕa + DЕк + Еэл +Еконт, (83)
где Еа и Ек - равновесные потенциалы анода и катода; DЕa и DЕк - анодная и катодная поляризация; Еэл и Еконт - падение напряжения в электролите и контактах. В ряде случаев в Uпракт входят и другие потери: падение напряжения в диафрагме (в случае разделения катодного и анодного пространств) и т. д. При электролизе стремятся уменьшить напряжение на ячейке за счет величин поляризации и омических составляющих баланса напряжения, так как напряжение разложения (Еa – Ек) изменить нельзя.
Падение напряжения в электролите зависит от его сопротивления R=xL/s, где х -удельное сопротивление электролита, ом . см; L - расстояние между электродами; s - площадь сечения электролита, через которую проходит электрический ток, см1. В случае неравных площадей анода и катода s=Ösasк.
Величину Еэл можно уменьшить сближением электродов, введением в раствор более электропроводящих добавок, повышением температуры.
Примерный электрический баланс электролизера на 30 000 а для получения металлического натрия электролизом расплава NaCl приведен в табл. 37.
Таблица 37
Электрический баланс электролизера на 30 000 а (плотность тока на катоде 0,8-0,95, на аноде 0,9-1 а/см2)
| Составляющие баланса | Падение напряжения | |
| в | % | |
| Напряжение разложения | 3,47 | 46,5 |
| Падение напряжения в электролите | 2,00 | 25,7 |
| Падение напряжения в аноде (включая контакты) | 0,90 | 12,0 |
| Падение напряжения в катоде и токоподводах (включая контакты) | 0,48 | 7,4 |
| Остальные потери (в шинах и т. п.) | 0,62 | 8,4 |
| Всего | 7,47 | 100 |
Обеспечив подачу на электроды необходимого напряжения, можно осуществлять электролиз с заданной скоростью. Повышение приложенного напряжения приводит к увеличению тока, протекающего через электролизер, - к повышению скорости процесса.
Количество выделяемого металла определяется законом Фарадея:
q = ZIt, (84)
где q - количество вещества, получаемое на электроде, г; I - ток, а; t - время, ч; Z-электрохимический эквивалент, равный количеству граммов вещества, выделяемого при протекании через электролизер тока I а в течение 1 ч.
Электрохимические эквиваленты некоторых металлов приведены в табл. 38.
Таблица 38
Плавкость солевых систем
Для успешного проведения электролиза расплавленных солей в большинстве случаев важно иметь электролиты с минимальной температурой плавления. Температура плавления связана с энергией решетки соли.
Энергию решетки соединений, имеющих ионы типа инертного газа, приблизительно рассчитывают по уравнению А. Ф. Капустинского:
U = 256(n+ + n-)/rк + ra ккал/г-экв,
где n - валентность; r - радиус, А.
Чем выше энергия кристаллической решетки, тем выше должна быть температура для расплавления соли. Следовательно, для соединений, описываемых формулой Капустинского, температура плавления падает по мере роста суммы радиусов катиона и аниона (табл. 39).
Таблица 39
Зависимость температуры плавления галогенидов металлов I и II групп от радиуса ионов
| Катион | rк, Å | Температура плавления, °С | |||
| F-, 1,33 Å | Cl-, 1,813 Å | Br-, 1,96 Å | I-, 2,20 Å | ||
| Li+ | 0,78 | 870 | 614 | 549 | 443 |
| Na+ | 0,98 | 997 | 800 | 755 | 651 |
| K+ | 1,33 | 846 | 790 | 734 | 680 |
| Rb+ | 1,49 | 780 | 715 | 681 | 642 |
| Cs+ | 1,65 | 683 | 646 | 610 | 600 |
| Be+2 | 0,34 | 800 | 404 | ||
| Mg+2 | 0,78 | 1270 | 718 | ||
| Ca+2 | 1,06 | 1478 | 772 | ||
| Sr+2 | 1,27 | 1190 | 868 | ||
| Ba+2 | 1,43 | 1280 | 958 | ||
Для фторидов, бромидов и хлоридов Cs-Na указанная закономерность выполняется. Падение температуры плавления для солей лития связано с большой поляризующей способностью лития, и частичной ковалентностью связи. Для иодидов падение температуры плавления начинается с натрия, так как даже ион Na+ способен поляризовать большой анион I-.
Увеличение доли ковалентной связи приводит к увеличению сил притяжения внутри каждой пары ионов и уменьшению этих сил между парами (направленность ковалентной связи), что и приводит к снижению прочности кристалла.
Для металлов второй группы увеличение суммы радиусов ионов приводит к повышению температуры плавления, так как двухзарядные ионы оказывают сильное поляризующее действие на анионы, а чем меньше радиус иона, тем большее поляризующее действие он оказывает (хлорид бериллия, например, в расплавленном состоянии не проводит ток, что подтверждает ковалентный характер связи). Увеличение радиуса двухвалентного иона уменьшает его поляризующую способность (особенно по отношению к F-), поэтому повышение температуры плавления в ряду двухвалентных ионов прекращается у CaF2. Для хлоридов (С1- поляризуется сильнее F-) зависимость температуры плавления от суммы радиусов имеет монотонный характер'.
По периодам периодической системы температура плавления солей меняется следующим образом:
| NaF | 997° С | AlF3 | Возгоняется без плавления |
| MgF2 | 1360° С | SiF4 | -77° С |
Вначале уменьшение радиуса катиона и увеличение его заряда приводят к упрочнению решетки соли и повышению температуры плавления, которая затем уменьшается вследствие роста доли ковалентности связи.
Для металлов, имеющих несколько степеней окисления, уменьшение валентности приводит чаще всего к понижению ковалентности связи и повышению прочности решетки (и температуры плавления):
| UF6 | 64,05° С | SnCl4 | 30,2° С | PbCl4 | 15° С | TiCl4 | 30° С |
| UF5 | 348° С | SnCl2 | 247° С | PbCl2 | 501° С | TiCl2 | 400° С |
| UF4 | 1036° С |
Окислы большинства металлов имеют ионные решетки, что обусловливает высокую температуру плавления. При плавлении смеси двух солей температура плавления зависит от состава смеси. Рассмотрим наиболее простые типы диаграмм плавкости двойных систем. На рис. 75 представлены три простейших типа таких диаграмм. Диаграммы плавкости описывают равновесия в гетерогенных системах.
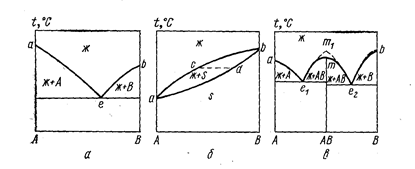
Рис. 75. Простейшие типы диаграмм плавкости двойных систем (Баймаков Ю. В., Ветюков М. М., 1966, с. 43, рис. б):
a- эвтектический тип; б- непрерывный ряд твердых растворов; в- с конгруэнтно плавящимся химическим соединением.
Выражение правила фаз [уравнение (40)] для солевых систем имеет вид
F = k - ф + l.
На диаграмме эвтектического типа (см. рис. 75, а) над линией ликвидиуса аеb имеется раствор веществ А и В друг в друге (одна фаза, два компонента, F = 2). Эта область дивариантна, в ней можно произвольно менять оба параметра - температуру и состав. На линиях ае и be жидкость находится в равновесии с кристаллами А и В - система моновариантна (изменив состав для сохранения числа фаз, необходимо двигаться по линии ае или be). В эвтектической точке е, когда в равновесии находятся три фазы – жидкость + А + В, система становится нонвариантной - нельзя произвольно менять ни состав, ни температуру без нарушения равновесия.
В случае неограниченной взаимной растворимости компонентов в жидком и твердом состояниях (см. рис. 75,6) система моновариантна. Раствор, состав которого выражается, например, точкой с, находится в равновесии с гомогенным твердым раствором, состав которого выражается точкой d. При кристаллизации смеси состава с фигуративная точка жидкой фазы движется от с к а, а твердой фазы - от d к a.
Если компоненты А и В обладают неограниченной растворимостью в жидком состоянии и образуют химическое соединение АВ в твердом состоянии, на линии ликвидуса должны быть три ветви, пересекающиеся между собой: две ветви кристаллизации чистых компонентов и ветвь кристаллизации химического соединения. При этом возможны два случая: состав жидкой фазы, образующейся при плавлении химического соединения, совпадает с составом его в твердом виде - конгруэнтное плавление (соединение АВ называется конгруэнтным) или состав соединения не совпадает с составом жидкой фазы, образующейся при его плавлении, - инконгруэнтное плавление. В случае конгруэнтного плавления на диаграмме плавкости соединению АВ отвечает максимум (дистектака), а диаграмму можно разбить на две части А-АВ и АВ-В, которые идентичны простым эвтектическим диаграммам (см. рис. 75, в). Обычно при плавлении химического соединения происходит частичный термический распад: АВ«А+В. Чем больше степень распада, тем больше радиус кривизны линии ликвидуса в дистектике. При отсутствии распада над химическим соединением линия ликвидуса претерпевает излом (точка m1).
В случае инконгруэнтного плавления (плавление малопрочных соединений) на диаграмме плавкости имеется «скрытый» максимум.
Полная взаимная растворимость в жидком и твердом состояниях (см. рис. 75,б) свидетельствует о близости структуры солей. Обычно такой тип диаграммы наблюдается для смесей солей чисто ионного строения с общим анионом или в том случае, если катионы близки по своим поляризационным свойствам (поляризующая сила и поляризуемость). Примеры системы: NaCl-KCl; Na2SO4-Li2SO4 и т. д.
Если кристаллические решетки индивидуальных солей смеси различаются значительно, возможны случаи полного смешения в жидком состоянии и раздельная эвтектическая кристаллизация в твердом.
Когда в смеси АХ-ВХ один из катионов (например, В"+) обладает большим зарядом и меньшим радиусом, силы связи Вn+—Х- будут больше, чем А+—Х-. Ион Вn+ выступает в качестве комплексообразователя, и образуется достаточно прочная группировка ВХm(m-n)-, в то время как катион А+ остается относительно свободным. Комплексообразование проявляется тем отчетливее, чем больше различие в поляризационных свойствах катионов. Диаграмма плавкости такой системы часто имеет вид, представленный на рис. 75, в.
Вязкость
Коэффициентом вязкости или внутреннего трения называется коэффициент пропорциональности в уравнении Ньютона для силы трения между двумя слоями жидкости, движущимися параллельно и ламинарно:
f = hF dw/dx, (86)
где f - сила трения, н; h - вязкость; F - площадь соприкосновения слоев, м2; dw/dx - градиент скорости между слоями по нормали к направлению потока, сек-1.
Вязкость обычно выражается в пуазах (1 пз=1 дин.сек/см2 =0,1 н.сек/м2}. Вязкость воды при 20° С и 1 ат равна 1,0087 спз. Величина, обратная вязкости, называется текучестью.
В техническом электролизе мы имеем дело с движением жидкости относительно неподвижных электродов, с перемещением капель металла и пузырьков газа в электролите. Для качественной и количественной характеристик этих процессов необходимо знать вязкость электролита. Вязкость расплава LiCl при температуре 617°С равна 1,81 спз, КС1 (t = 790°С) - 1,42 спз.
Вязкость расплавленных солей падает с ростом температуры. Температурная зависимость вязкости имеет вид
h= Aexp(Eh/kT), (87)
где Eh - энергия активации вязкого течения, ккал/моль; k - константа Больцмана, ккал/град; Т - абсолютная температура.
Считается, что вязкость смеси определяется наиболее громоздкими частицами, так как их переход в соседнее равновесное положение—самый медленный процесс (наибольшая энергия активации). Анионы, как правило, превосходят по размерам катионы, а расплавы соли со сложными анионами (например, Сr2О72- и т. п.) имеют повышенную вязкость. Повышается вязкость и в системах, где возможно образование сложных частиц типа автокомплексных ионов (MgCl2, CdCl2, CaCl2 и т. п.).
Вязкость - свойство не аддитивное, и для идеальных систем изотермы вязкости являются монотонными кривыми, выпуклыми к оси состава. Химическое взаимодействие компонентов в ионных системах приводит к укрупнению ионов (комплексные ионы), при этом на изотермах вязкости можно ожидать резких максимумов или точек перегиба. Появление в гомогенной жидкости взвешенных твердых частиц приводит к резкому возрастанию вязкости.
Поверхностные явления
Образование и рост кристаллов на электроде, образование капель жидкого металла на катоде, всплывание металла в электролите, растворение металла в расплавленных солях, вопросы смачивания, пропитывания солями футеровки электролизера - все эти процессы во многом определяются межфазным натяжением, существующим на соответствующей границе раздела фаз.
Частицы жидкости, находящиеся в поверхностном слое на границе с газом, притягиваются, внутрь жидкости. Эта поверхностная пленка стремится сжать жидкость, поэтому при увеличении поверхности жидкости приходится затрачивать работу. Эта работа, отнесенная к 1 см2 новой поверхности жидкости, называется поверхностным натяжением о и измеряется в эрг/см2.
Поверхностное натяжение мало зависит от природы газа, с которым соприкасается жидкость, так как силы взаимодействия между частицами жидкости намного больше, чем силы, действующие между частицами жидкости и газа. Рост температуры приводит к уменьшению сил взаимодействия между частицами жидкости и к уменьшению поверхностного натяжения:
st = a - b{t - tпл), (88)
где st - поверхностное натяжение при температуре t °C; tпл - температура плавления; а и b - постоянные для данной соли.
Рост радиуса катиона в ряду солей с одноименным анионом приводит к падению сил межионного притяжения и, .следовательно, к уменьшению а. Однако рост радиуса катиона может приводить к уменьшению взаимной поляризуемости катиона и аниона, уменьшению доли ковалентности связи и, следовательно, к росту сил межионного притяжения и росту s.
Для идеальных смесей изотерма поверхностного натяжения представляет собой кривую с небольшой выпуклостью к оси состава.
Естественно, что чем больше напряженность собственного поля частицы или чем больше ее обобщенный момент ne/r, тем прочнее она удерживается окружающими ионами. Следовательно, частицы с меньшим обобщенным моментом должны «выталкиваться» в поверхностный слой, где их концентрация больше, чем во всей массе жидкости. Такие вещества называются поверхностно-активными, и с повышением их концентрации в пограничном слое поверхностное натяжение падает. Образование комплексных частиц должно приводить к падению величины а, так как образующиеся частицы имеют значительные размеры и малый обобщенный момент и должны быть поверхностно-активными.
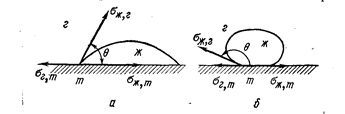
Рис. 76. Схема сил межфазного натяжения, действующих на частицу жидкости, лежащую на границе трех фаз (Баймаков Ю. В., Ветюков М. М., 1966, с. 43, рис. б):
a - жидкость хорошо смачивает твердое тело; sг,ж > sж,т; cosq >0; б - жидкость плохо смачивает твердое тело; sг,ж < sж,т; cosq <0.
Если нанести каплю жидкости на ровную поверхность твердого тела, то жидкость либо смачивает поверхность и растекается, либо плохо смачивает и сжимается в каплю. Частица жидкости, лежащая на границе трех фаз, подвержена действию трех сил межфазного натяжения на границах (рис. 76): газ - твердое (sг,т), жидкое-твердое (sж,т) и жидкое-газ (sж,г). Условие равновесия частицы - равенство проекции сил на горизонтальную линию:
sг,т = sж,т + sж,гcos q (89)
где q - краевой угол смачивания; cos q = (sг,т - sж,т) / sж,г.
В табл. 40 приведены данные по смачиванию поверхности угля расплавами некоторых солей.
Таблица 40
Диффузия
Самопроизвольный перенос вещества, причиной которого является хаотическое тепловое движение частиц, называется молекулярной диффузией.
Конвективная диффузия связана с переносом микрообъемов жидкости под действием перемешивания, вызванного разностью температур или удельных объемов внутри жидкости.
Здесь рассмотрим лишь молекулярную диффузию.
Очевидно, что диффузия пропорциональна скорости движения частиц (скорости перескока в соседнее положение). Такой перескок характеризуется энергией активации. Средняя скорость движения частиц
w = d/t,
где d - расстояние между частицами; t - среднее время оседлой жизни частицы:
t = t0 exp(Ed/RT)
где Ed - энергия активации диффузионного перехода; D - коэффициент диффузии, обратно пропорционален t; D = Doexp(-Ed/RT). Следовательно, коэффициент диффузии растет с ростом температуры по экспоненциальному закону.
Величины D жидких металлов и солей друг в друге имеют порядок 10-5 см2/сек.
Электропроводность и перенос ионов в расплавленных солях,
Удельная электропроводность есть величина, обратная удельному сопротивлению c = 1/r ом-1.см-1. Если между двумя параллельными электродами, находящимися на расстоянии 1 см друг от друга, поместить 1 моль или.1 г-экв вещества, то получаемая величина электропроводности называется молекулярной (m) или эквивалентной (l}:
m = cV = cM/r ом-1 .см2;
l = cV/m ом-1 .см2,
где M - молекулярная масса соли, г; V - мольный объем соли, cм3/моль; r - плотность соли, г/см3; m - число грамм-ионов одного знака, на которые распадется грамм-моль соли при диссоциации.
В табл. 42 приведены эквивалентные электропроводности некоторых расплавленных солей. Важно при выборе состава электролитов подбирать наиболее электропроводные смеси, поскольку при этом возможно повысить силу тока без нарушения теплового режима электролизера.
Таблица 42
Эквивалентная электропроводность, ом -1 . см2, хлористых соединений при температуре плавления
| Группа периодической системы | |||||
| I | II | III | IV | V | VI |
| HCl 10-6 | |||||
| LiCl 166 | BeCl2 0,086 | BCl3 0 | CCl4 0 | ||
| NaCl 133,5 | MgCl2 28,8 | AlCl3 1,5.10-5 | SiCl4 0 | PCl5 0 | |
| KCl 103,5 | CaCl2 51,9 | ScCl3 15 | TiCl4 0 | VCl5 0 | |
| RbCl 78,2 | SrCl2 55,7 | YCl3 0,5 | ZrCl4 - | NbCl5 2.10-7 | |
| CsCl 66,7 | BaCl2 64,6 | LaCl3 29,0 | HfCl4 - | TaCl5 3.10-3 | MoCl6 1,8.10-6 |
| ThCl4 16 | WCl6 2.10-6 | ||||
Увеличение электропроводности связано с падением поляризующей силы катиона. Связь Ве-Сl практически нацело ковалентна, расплав BeCl2 почти не проводит тока.
Электропроводность связана с вязкостью, поскольку вязкость среды определяет сопротивление движению иона. Оказалось, что электропроводность растет с температурой значительно медленнее, чем падает вязкость, и в большинстве систем хорошо выполняется уравнение lKh = const. При этом постоянная К колеблется от 2,8 до 1,8 для различных солей.
Изотерма электропроводности смесей, близких к идеальным, представляет собой кривую, выпуклую к оси состава. В случае образования соединений наблюдаются отклонения от идеальной изотермы: минимумы (из-за громоздкости комплексных ионов), S-образование изотермы (когда электропроводность компонентов смеси неодинакова), небольшие максимумы, когда компоненты в смеси ассоциированы в меньшей степени, чем чистые компоненты.
ПРОМЫШЛЕННОЕ ОФОРМЛЕНИЕ ЭЛЕКТРОЛИЗА
Цех электролиза
Электролизеры питаются постоянным током от выпрямителей - ртутных или полупроводниковых.
Электролизеры с расплавленными солями выделяют в окружающее пространство большое количество тепла. Кроме того, несмотря на герметизацию или местный отсос, в помещение цеха проникают анодные газы. Это создает вредные условия работы. Часовой обмен воздуха в цехе должен быть равен 10-30-кратному. Внутри цеха отделывают кислотостойкой краской. Токопроводящие шины и вентиляционные каналы, трубопроводы для подачи воды или воздуха обычно располагаются в подвальном помещении.
Специфика работы электролизера делает необходимым соблюдение особых правил техники безопасности. Высокая реакционная способность металлов и солей не позволяет находиться в цехе без спецодежды (куртка и брюки или комбинезон из огнеупорной ткани, перчатки, ботинки с покрытиями, очки, головной убор). У аппаратчиков электролизного отделения в случае выделения на аноде вредных газов (Cl2, F2 и т.п.) должен быть при себе исправный противогаз в положении наготове. Все работающие должны уметь оказывать первую помощь пострадавшему при ожогах и поражениях током.
В цехах электролиза принимают специальные противопожарные меры. Ни в коем случае нельзя пользоваться углекислотными огнетушителями или хлорированными углеводородами при тушении пожаров. Огонь нужно гасить, засыпая горящий металл большим количеством твердого инертного материала - сухим хлористым натрием и т.п.
Легковоспламеняющиеся металлы хранят под слоем масла (Li, другие щелочные металлы) или под водой (порошки Ti, Zr, Hf и др.).
Тугоплавкие металлы
Все тугоплавкие металлы (за исключением бериллия) являются переходными элементами. Им свойственны образование катионов нескольких валентностей, высокая стойкость в компактном состоянии и высокая химическая активность в тон-кодисперсном состоянии или при повышении температуры. При нагревании большинство из них активно реагируют с кислородом, азотом и водородом.
К группе тугоплавких относятся (в скобках указана температура плавления металлов, °К): Be (1558°); Sc (1811°); Y (1782°); Ti (1941°); Zr (2125°); Hf (2495°); V (2173°); Mb (2741°); Та (3269°); Cr (2148°); Мо (2883°); W (3683°) Re (4353°); Th (2023°).
Все тугоплавкие металлы (кроме Be) образуют галогенидные соединения высшей валентности, которые в контакте с металлом (а для некоторых - с водородом) при высокой температуре восстанавливаются до соединений низшей валентности.
Таблица 46
Температура плавления и кипения некоторых галогенидов тугоплавких металлов
| Галогенид | tпл, °К | Tкипo K, при которой р'МеС1n= 760 мм pm. cm. | Галогенид | tпл, °К | Tкипo K, при которой р'МеС1n= 760 мм pm. cm. |
| BeCl2 | 678 | 488 | MoCl5 | 467 | 541 |
| YCl3 | 953 | — | WCl6 | 548 | 619,7 |
| TiCl4 | 250 | 409,5 | WCl5 | 521 | 548,6 |
| TiCl3 | 1003 | 1023-1123 (возг.) | ВеF2 | 1080 | 1073 (возг.) |
| TiCl2 | 1298 | 1788 | TiF4 | — | 557 |
| ZrCl4 | 710 (под давл.) | 604-903 (возг.) | ZrF4 | (1200) | (1200) (возг.) |
| ZrCl3 | (900) | — | ZrF3 | (1600) | (2373) |
| ZrCl2 | (1000) | (1753) | ZrF2 | (1800) | 2473 |
| HfCl4 | 705 (под давл.) | 590 | HfF4 | (1203) | (1173) |
| ThCl4 | 1093 | 1192 | NbF5 | 353 | 507,9 |
| VCl4 | 247 | 425 | TaF5 | 368,1 | 502,2 |
| NbCl5 | 482,5 | 527,0 | MoF6 | 290 | 308 |
| ТаCl5 | 493,0 | 512,3 | WF6 | 275,5 | 292,5 |
Примечание. Числа в скобках - ориентировочные данные.
Галогенидные соединения высшей валентности тугоплавких металлов в основном имеют ковалентную связь, практически не электропроводны, плавятся и кипят при низкой температуре (табл..46).
С галогенидами щелочных и щелочноземельных металлов образуются соединения типа NanMeXm+n, в которых тугоплавкие металлы входят в комплексный анион. Фторидные комплексы наиболее прочны. При разряде катионов тугоплавких металлов на катоде образуются кристаллы металла, так как температура электролита в этом случае всегда ниже температуры плавления выделяемого металла.
Широкое внедрение в технологию редких металлов хлорного метода переработки рудных концентратов позволяет получить достаточно чистые хлориды (применяя метод раздельной конденсации), которые после дополнительной очистки возгонкой, ректификацией и т. и. являются исходными соединениями для получения металлов электролизом.
Вследствие ковалентности связи большинство хлоридов тугоплавких металлов слабо проводят электричество. Кроме того, сравнительно низкая температура кипения (или возгонки) определяет высокое давление пара хлоридов при температуре электролиза. Поэтому электролиз расплавов чистых хлоридов тугоплавких металлов практически невозможен. Электролиз ведут из их смеси с КСl или NaCl. При этом образуются химические соединения, понижающие давление пара хлоридов над расплавом. Обычно в электролите концентрацию хлоридов тугоплавких металлов поддерживают 3-12 мол. %, а температуру 950-1200° К. Концентрация хлоридов тугоплавких металлов должна быть достаточной, чтобы не допустить возникновения концентрационной поляризации на катоде, что может вызвать разряд посторонних ионов, снизить выход по току и чистоту получаемого металла.
Материалом анода обычно служит графит. В случае электролитического рафинирования анод изготовляют из блока очищаемого металла или металл в виде стружки и обрезков помещают в сетчатые корзины, располагаемые вокруг катода (металл сетки заметно электроположительнее очищаемого металла). Катодами обычно служат цилиндрические стержни различных диаметров либо металлические пластины. Металл катода не должен реагировать с расплавом и металлом, на нем осаждаемым. Чаще всего применяют стандартные жаропрочные стали. Нередко катодом служат стержни или пластины того металла, который подвергается в данном случае электролитическому осаждению.
Электролизер простейшего типа состоит из стального котла, вставленного в печь, обогреваемую электрическим током или газом. В котел помещается стальной стакан, в котором и происходит процесс электролиза (стакан при этом служит катодом). Котел герметически закрывается крышкой, в центре которой находится графитовый анод. В крышке имеются также патрубки для отвода хлора, подачи аргона под давлением и слива электролита под давлением. Электролит по мере обеднения пополняют солью металла. По окончании цикла электролит сливают, электролизер разбирают, стакан вытаскивают, осадок промывают горячей водой (или слабой кислотой) и снимают со стенок. Процесс в таком электролизере периодический.
Часто используют открытые электролизеры непрерывного действия. При этом процесс осуществляется на воздухе, что может существенно сказаться на качестве получаемого металла. Стальной котел электролизера (рис. 78) охлаждают водой, вследствие чего на стенках и днище образуется гарниссаж из соли, не допускающей реакции материала стенок с расплавом. Нужную температуру поддерживают переменным током, подаваемым на пару электродов. В электролизер заливают расплавленный электролит, после чего включают ток - переменный и постоянный. Хлор удаляют посредством бортового отсоса. На стержнях-катодах образуется масса, состоящая из металлических кристаллов различной крупности и электролита («груша»). Электролизер питают твердой солью.
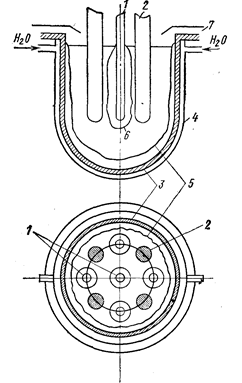
Рис. 78. Схема электролизера непрерывного действия (Ваймаков Ю. В., Ветюков М. М. 1966, с. 516, рис. 157):
1-стальные или молибденовые катоды; 2-графитовые аноды: 3-корпус ванны (сталь); 4-кожух водяного охлаждения; 5- гарниссаж из электролита: 6 - катодный осадок; 7 - бортовой отсос.
Катодных стержней обычно несколько (например, четыре анода и пять катодов). По мере образования осадка катодные стержни поочередно извлекают из расплава, процесс электролиза при этом не прерывается. Извлеченную «грушу» обжимают прессом над ванной для удаления из нее части электролита. Осажденный металл всегда содержит заметное количество азота и кислорода.
Для предотвращения контакта металла с воздухом предложена конструкция герметического электролизера непрерывного действия (рис. 79).
Компоненты электролита переплавляют в вакууме и хранят в герметичной таре. Перед пуском электролизер заливают электролитом, включают переменный ток, откачивают воздух и промывают аппарат аргоном. Электролизер может быть снабжен патрубками для спуска электролита и шлама.
Современные требования к чистоте получаемых тугоплавких металлов очень высоки, поэтому применяют только специально очищенные и перекристаллизованные в перегнанной воде соли. Аргон и гелий для промывки аппаратов очищают сорбцией на глиноземе, пятиокисью фосфора и сорбцией на губчатом титане при 800° С.
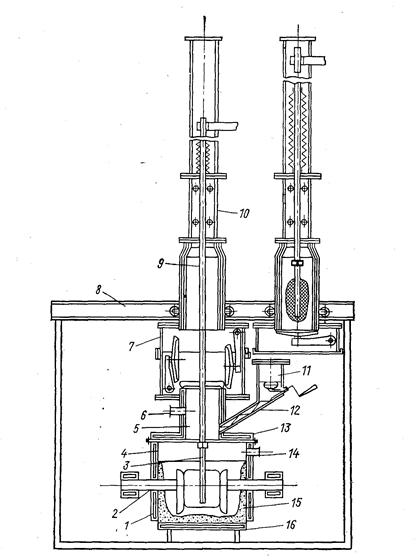
Рис. 79. Схема герметичного электролизера со сменными катодами (Баймаков Ю. В., Ветюков М. М., 1966, с. 517, рис. 158):
1 - стальные водоохлаждаемые стенки; 2 - четыре графитовых анода (из них два питаются переменным током для поддержания нужной температуры в электролите); 3 - никелевый катод, присоединенный к полому водоохлаждаемому стальному катодо-держателю; 4 - сварной герметизированный корпус электролизера; 5 - переходный патрубок на крышке; 6 - патрубок для удаления газов, ванна имеет патрубок для спуска электролита и шлама; 7 - разъемный шлюз с крышками, закрывающими электролизер н камеру, шлюз двигается на роликах; 8 - рама для передвижения камер; 9 - катододер-жатель; 10 - камера для приема и охлаждения осадка без доступа воздуха; 11 - герметичный загрузочный бункер с вакуумными затворами; 12 - питатель для подачи в ванну свежего электролита и корректирующих добавок; 13 - водоохлаждаемая крышка на ванне с резиновой прокладкой и охлаждением; 14 - патрубок для подачи инертного газа и для откачки воздуха; 15 - гарниссаж на стенках и днище ванны; 16 - стальное водоохлаждаемое днище.
Бериллий
Электролитическое восстановление бериллия из водных растворов невозможно, так как его галогенидные соли подвержены гидролизу и ионы сильно гидратированы. Используют расплав состоящий из BeCl2-NaCl. Эвтектика с содержанием 50 мол % BeCl2 плавится при 215° С. Потенциал разложения BeCl2 E =2,04-0,00055 (T-773° К) в, а в эквимолярнои смеси E873 K =1,99 в; E973°K = 1,73 в. Потенциал разложения зависит от концентрации BeCl2 в расплаве, Е773 К = 1,962 + 0,244/N2 , где N - мольная доля BeCl2.
В промышленных условиях в качестве электролита используют смесь BeCl2 - (NaCl + KCl) в отношении 1:1. Электролиз проводят в электролизерах периодического действия. Плотность тока на аноде 0,37 а/см2, на катоде 0,08 а/см2, сила тока до 2000 а, напряжение на ванне 4-6 в. Выход по току 70-80% удельный расход энергии 32-50 квт. ч/кг. В расплаве поддерживают концентрацию BeCl2 5-7 масс. %. Кристаллы бериллия отмывают от солей, раствор солей направляют на регенерацию (с получением Be(ОН)2). Используя сменные катоды можно очищать электролит предварительным электролизом, при этом осаждаются более электроположительные примеси, и катод заменяют на новый для осаждения бериллия.
Чистоту металла, получаемого в результате электролиза, характеризуют следующие данные по примесям, масс. %:
| B | 1.10-5 | Al | 3.10-3 | Ca | 1,4.10-3 |
| Li | 1.10-5 | Si | 2,4.10-2 | Cl | 2,2.10-3 |
| Ag | 1.10-4 | Cd | 6.10-6 | S | 1.10-2 |
| Mn | 2,3.10-3 | Co | 1.10-4 | Sn | 5.10-3 |
| Cu | 1,7.10-3 | Ni | 3.10-3 | P | 1.10-2 |
| Fe | 7.10-3 | Zn | 1,6.10-3 | F | 5.10-2 |
| Mg | 1.10-3 | Ti | 1.10-3 | Pb | 1.10-2 |
| Ba | 6,6.10-3 | Cr | 8.10-4 |
Металл такой чистоты не удовлетворяет требованиям, предъявляемым специальным машиностроением. В настоящее время бериллий высокой чистоты получают из особо очищенной окиси бериллия, прохлорированной и возогнанной в вакууме Электролиз ведут в герметичном электролизере с непрерывной подпиткой BeCl2 из герметичного бункера. Получающийся металл дополнительно очищают электролитическим рафинированием - проведением электролиза в тех же условиях с расходуемым анодом.
Наиболее существенными недостатками электролитического процесса являются: корродирование электролизера, большое давление пара хлоридов при температуре электролиза и трудность отделения бериллия от расплава.
Возможен электролиз расплава фторида бериллия. В этом случае в качестве электролита, применяют NaF- BeF2 или BaF2-BeF2. Электролиз смеси NaF-BeF2 . проводят при температуре около 600° С с получением твердых кристаллов металла, смеси BaF2-BeF2 - при температуре 1400° С с получением расплавленного металла. Катодом служит никелевый или железный стержень. Однако применение для электролиза фторидов не решает проблемы летучести электролита, коррозия электролизеров при этом даже увеличивается. С другой стороны, образование во фторидной системе прочных комплексов бериллия снижает выход по току вследствие частичного выделения металлов-примесей. Электролиз фторидов в настоящее время в промышленности не применяют.
Металлический бериллий получают в основном металлотермическим восстановлением фторидов. Удельный вес электролиза в общем объеме производства металла составляет всего ~15%. Одной из причин этого является высокая летучесть хлоридов, которые могут образовывать весьма ядовитые аэрозоли.
Титан.
Для электролиза используют галогенидные соединения титана, преимущественно хлориды, растворенные в расплавах NaCl и КСl. Растворимость TiCl4 в LiCl и КСl доходит до 1,5 масс.%. В CsCl она возрастает до 6%. Добавки NaF повышают растворимость до 2-4% (в 10-20%-ном NaF). Повышение растворимости объясняется образованием комплексной соли Na2TiFe.
Низшие хлориды титана растворимы в расплавленных хлоридах калия и натрия с образованием комплексных соединений: К3ТiСl6 и KtiCl4; KTiCl3 и K2TiCl4.
Металлический титан реагирует с нонами высшей валентности, восстанавливая их:
3TiCl4 + Ti « 4TiCl3 lg K = - 4,09 + 10347/T;
TiCl4 + Ti « 2TiCl2 lg K = - 1,998 + 7968/T;
2TiCl3 + Ti « 2TiCl2 lg K = - 5,038 + 6774/T;
При 1000° К в расплаве NaCl-KCl, - содержащем 6,7 масс. % титана в виде хлорида, в равновесном состоянии с металлом на долю ионов Ti2+ приходится 95-96%, Ti3+- 4,1% и Ti4+- только 1.10-5%.
Установлено существование монохлорида титана, образующегося по реакции TiCl2+Ti « 2TiCl. Монохлорид существует при повышенной температуре и чрезвычайно нестоек. Образование и распад монохлорида вызывают появление дисперсной металлической фазы в расплаве.
Существуют два метода электролитического получения титана: 1) электролиз TiCl4 с применением графитового анода; 2) электролитическое рафинирование титана в расплавах низших хлоридов с расходуемым анодом.
Добавление в электролизер летучего хлорида — технически трудно осуществимый процесс. Обычно в этом случае применяют электролизеры с двойным дном (схема подачи летучего хлорида представлена на рис. 80).
На катоде происходит восстановление до TiCl3 и TiCl2 (молекулы низших хлоридов диффундируют в расплав) и восстановление Тi3+ и Ti2+ до металла. На аноде происходит окисление анионов хлора. Выход по току (по хлору) равен 94%. Наиболее благоприятная температура 800° С. Анодная плотность тока 0,8 a/cм2. Понижение анодной плотности тока приводит к уменьшению анодного выхода по току, при повышении возможно возникновение анодного эффекта.
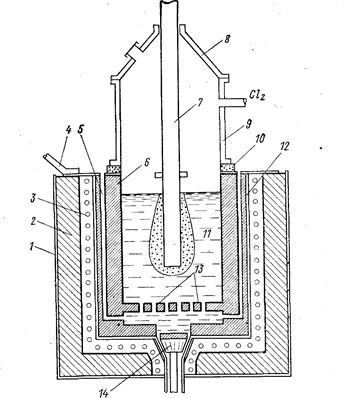
Рис. 80. Схема электролизера для электролитического получения тугоплавкого металла с подачей легкокипящих жидких хлоридов (Баймаков Ю. В., Ветюков М. М., 1966, с. 518, рис. 159):
1 - наружный кожух; 2 - теплоизоляция; 3 - электронагреватели; 4 - анодный токопод-вод; 5 - вертикальные каналы для подачи жидкого хлорида; 6 - цилиндрический графитовый анод; 7 - катод из жаропрочной стали; 8 - съемный колпак; 9 - верхний кожух из жаропрочной стали; 10 - электроизоляция; 11 - электролит; 12 - внутренний кожух из жаропрочной стали; 13 - ложное днище с отверстиями для подачи испаряющегося хлорида; 14 - пробка для спуска электролита.
При повышении плотности тока на катоде выделяется щелочной металл или его субион (Na2+), который восстанавливает TiCl4 до TiCl3 и TiCl2. Низшие хлориды на катоде восстанавливаются до металла.
В настоящее время применяют расплавы состава 50% NaCl - 30% KCl -10% NaF. Электролиз ведут при 680-700° С, плотность тока на катоде достигает 8 а/см2, при этом выход по току ~ 70%, напряжение на ячейке 6 в. Металл содержит некоторое количество углерода (из анодов), и при недостаточной герметизации аппаратуры может иметь место повышенное содержание кислорода и азота. Катодный осадок состоит из чрезвычайно мелких кристаллов, так как на катоде протекают реакции поляризации ионов, нарушающие рост кристаллов.
Однако подбором оптимального сочетания катодной и анодной плотностей тока, удельной скорости подачи TiCl4 и конструкции электролизера (взаимного расположения анода и катода) можно достигнуть такого сочетания протекающих реакций, при котором соотношение TiCl3 и TiCl2 в расплаве обеспечило бы получение более крупных кристаллов.
Необходимость получения чистого титана, а также переработки некондиционной титановой губки, отходов титанового литья и механической обработки послужила толчком для разработки процесса электролитического рафинирования металла.
Элетролитическое рафинирование проводят в герметически закрытых электролизерах, заполненных аргоном. Куски губки, стружку и т. п. помещают в сетчатые стальные корзины, которые служат анодом. Электролитом служит расплав NaCl- TiCl3 – 8-15 масс. % TiCl2 при температуре 850-900° С, а катодом - стальные стержни.
На титановом аноде в первую очередь идет реакция Ti®Ti2++2e-. По мере увеличения поляризации и плотности тока начинает идти реакция Ti®Ti3++3e-. Первая реакция идет при напряжении 2,1-2,2 в. При плотности тока на аноде 0,1 а/см2 практически образуются лишь ионы Ti2+, а при плотности тока 0,4-0,5 а/см2 ионы Ti2+ и Ti3+ переходят в расплав в равных долях.
На катоде электрохимические реакции идут в обратном порядке: Ti3++e-®Ti2+, Ti3++3e-®Ti, Ti2++2e-®Ti. По мере увеличения плотности тока последовательно начинают проходить три приведенные реакции; дальнейшее увеличение напряжения вызывает появление предельного тока и начало разряда ионов натрия.
При прохождении тока через ячейку электролитического рафинирования количество ионов титана, образующихся на аноде, не совпадает с их количеством, разряжающимся на катоде. Пересчет количества электричества, прошедшего через ячейку, на количество растворившегося и выделившегося металла позволяет определить средний заряд ионов, образующихся на аноде (2,03-2,1) и разряжающихся на катоде (2,5-2,3). Казалось бы, что в этих условиях расплав должен обогащаться ионами титана. Но на самом деле, напротив, расплав обедняется ионами титана. Это вызвано протеканием в расплаве реакции диспропорционирования 2TiCl2 ® Ti + TiCl4. Часть ионов Ti2+ уходит из расплава. В промышленных условиях эту убыль приходится компенсировать добавлением расплава NaCl – 40-50 масс. % (TiCl3- TiCl2).
Оптимальная исходная концентрация TiCl3 в расплаве 4-5 масс.%. Концентрация TiCl2 в процессе электролиза устанавливается 80% общего количества хлорида титана в расплаве. Анодная плотность тока 0,1-0,3 а/см2, катодная 1,8-3 а/см2, температура 800° С. Выход по току около 80% отвечает валентности 2.
Стальной катод сначала покрывается тонкой пленкой титана вследствие диспропорционирования TiCl2 и TiCl, затем осадок формируется в виде уплотненного скопления призмочёк. Размеры кристаллов увеличиваются с понижением плотности тока и повышением температуры. Кристаллизация улучшается в отсутствие ионов Ti3+. Понижение концентрации хлоридов титана в расплаве вызывает измельчение кристаллов. К такому же эффекту приводит присутствие в расплаве кислородных соединений, углерода, азота, ионов Аl3+, Mg2+, V4+, Ni2+ и некоторых других.
Получаемая очистка от железа, кремния, кислорода, водорода и азота удовлетворительна. Ионы некоторых примесей переходят в катодный осадок.
Электролит для рафинирования проще всего приготовить, пропуская пары TiCl4 через расплав NaCl, содержащий титановую губку при температуре 850° С. Таким образом готовят расплав с содержанием до 40% TiCl2- TiCl3Т1.
Аппаратуру изготовляют из стали марки Ст. 2. Электролизер обязательно имеет в днище отверстие для слива шлама с отработанным электролитом.
Осадок металла сбивают с катода, обрабатывают подкисленной дистиллированной водой, промывают, сушат в вакууме и рассеивают по крупности.
Электролизеры сооружаются на 10-30 ка. Напряжение на ванне 8-11 в. Выход по току колеблется от 60 до 85%. Расход энергии на 1 т металла 15000 кет-ч. Кроме того, на нагревание электролизера затрачивается 8000-10000 квт.ч переменного тока на 1 т металла.
Цирконий и гафний
Цирконий и гафний в расплавах образуют ионы Me4+, Me2+ и неустойчивые Ме+. Хлориды и фториды четырехвалентных металлов - ковалентные химические соединения, образующие с галогенидами щелочных металлов в расплавах комплексные соединения (такие, как Me2ZrF6, где Me=Na, К; Me3ZrF7 и т. п.).
Электрохимическим путем цирконий получают из расплавов смесей КСl-K2ZrF6 и NaCl- K2ZrF6. В этих расплавах в результате обменной реакции образуется комплексный анион ZrFeCl3-.
При взаимодействии иона циркония с металлом протекает реакция: Zr4++Zr«2Zr2+, lg K800oC = 10,2. В присутствии металла в расплаве преобладают ионы Zr2+.
Электролитическое получение технического циркония в настоящее время осуществляют в открытых электролизерах на ток 2000-6000 а.
'На аноде окисляется ион хлора. Суммарная реакция на электродах K2ZrF6 + 4KCl ± e- « Zr + 2Cl2 + 6KF.
Во время электролиза в электролит периодически вводят порции соли K2ZrF6-КС1. В электролите должны накапливаться ионы F+ и К+. Однако содержание фтора в электролите балансируется тем, что значительное количество расплава с повышенным содержанием KF захватывается катодным осадком, часть электролита уходит со шламом, выпускаемым периодически из ванны. На катоде образуется осадок, содержащий металл, электролит, низшие фториды циркония. Его подвергают дроблению, обрабатывают горячей водой, подкисленной НС1 (100 г/л), металл отмывают от шлама низших фторидов, сушат в вакууме при 60-100° С и рассеивают по фракциям.
Из сравнения данных по потенциалам разложения хлоридов циркония и гафния видно, что значение потенциалов для Hf/Hf4+ несколько электроотрицательнее (EoHfCl4 = 2,59-0,615.10-3 T в), чем для Zr/Zr4+ (EoЯкCl4 = 2,34 – 0,48.10-3 T в), однако для двухвалентных хлоридов картина обратная (EoHfCl2 = 2,51 – 0,4.10-3 T в; EoZrCl2 = 2,54 – 0,53.10-3 T в). Поэтому катионы Zr4+ и Hf4+ разряжаются одновременно. В смешанных фторидно-хлоридных растворах потенциал гафния отрицательнее потенциала циркония; разность при 700° С составляет 0,06 в, а при 800° С - 0,02 в. Термодинамически, следовательно, разделение на катоде возможно, однако при наложении кинетического фактора w многообразии катодных реакций раздельный разряд Zr4+- и Hf4+ становится маловероятным. Происходит лишь частичное обогащение катодного осадка по цирконию.
Электролитическое рафинирование циркония протекает аналогично рафинированию титана. При этом достигается хорошая очистка от кислорода, азота и водорода.
Для получения электролитического циркония достаточной. степени чистоты за одну операцию (без дополнительной очистки) процесс ведут в герметичном электролизере (см. рис. 79). Кристаллы K2ZrF6 дважды перекристаллизовывают и сушат в вакууме при 80° С.
Химически чистый КСl обезвоживают в муфельной печи при температуре 350-400° С. Состав расплава: 30-70% КСl, 10-30% K2ZrF6, 20-40% KF. Температура электролиза 750-800° С, катодная плотность тока 0,3-0,5 а/см2, выход пo току 50-60%. Продолжительность наращивания катодной «груши» 2-2,5 ч. В процессе электролиза для корректировки состава электролита в ванну вносят 20 г K2ZrF6 и 25 г КСl на каждые 10 а.ч, пропущенные через электролит. В открытых электролизерах чистота металла не превышает 0,3-0,4% по кислороду и 0,01% по азоту, а в герметичных электролизерах с защитной атмосферой содержание кислорода 0,06%, азота 0,003%. Катодный осадок содержит 30% металла и электролит.
Электрохимическое поведение гафния в основном подобно поведению циркония.
Ниобий и тантал
NbCl5, TaCl5, NbF5 и TaF5 образуют комплексные соединения типа K2MeF7. Фторниобат калия легко гидролизуется до K2NbOF5.Н2О. Фтортанталат более устойчив. При восстановлении образуются галогениды низших валентностей.
Компонентами электролита для получения обоих металлов служат КСl, K2MeF7, KF, в некоторых случаях используют окислы, растворяющиеся во фторидном расплаве. Значения потенциала разложения следующие:
EoNbCl5 = 1,94 - 0,50.10-3 Тв; EoТаCl5 = 2,47 (298 К) в;
EoNbCl3 = 1,76 - 0,467.10-3 Тв; EoNbCl5 = 2,0 (1023 К) в;
Процесс электролиза ведут с высокой катодной плотностью тока 1-2 а/см2, что позволяет достигнуть потенциала разряда ионов Me3+ и Me2+ и предотвратить их накопление и диспропорционирование.
При получении технических ниобия и тантала применяют открытые электролизеры. Материал катода - молибден. Состав электролита: 9-10% K2MeF7, 4-8-% Me2O5, 25-42% КС1, 44-57% KF.
Введение в электролит растворенных окислов улучшает смачиваемость анода и повышает критическую силу тока, выбывающую анодный эффект. Улучшение смачивания объясняется тем, что окислы в данном случае являются поверхностно-активными веществами. Добавка растворимых окислов в расплаве уменьшает краевой угол смачивания.
Температура электролиза 750° С, выход по току 80%. Чистота технического Та, масс. %: 0,1-0,2 О, 0,1-0,3 Fe+Ni, 0,01 F, 0,002 Mn, 0,003-0,1 С.
Для получения металлов повышенной чистоты применяют электролиты без Ме2О5 в герметичном электролизере (см. рис. 79). Состав электролита: 55% КСl, 27,5% KF, 17,5% K2MeF7. Температура 700-800° С, катодная плотность тока 0,6-0,8 а/см2, анодная 1,5-2,3 а/см2. Получающийся ниобий содержит, масс. %: 0,02 С; <0,05 О; 0,02 N.
Применяют в технологии и процесс электролитического рафинирования с растворимым анодом, приготовленным из металлического порошка методом металлокерамики. В данном случае применяют хлоридный электролит, состоящий из КСl, NaCl, МеСl3. Во фторидных расплавах ионы Nb5+ и Та5+ связаны в прочный комплекс, и для их выделения необходимо приложить большее напряжение, что влечет за собой совместный разряд, более электроотрицательных ионов (Al3+ и др.). Проведение электролиза в закрытых электролизерах улучшает чистоту получаемого металла. При этом электролит удаляют из катодного осадка вакуумной сепарацией при 1800-2000° С.
Редкоземельные металлы
Электролитом служат расплавы хлоридов и фторидов щелочных и щелочноземельных металлов. Более легкоплавкие редкоземельные металлы с температурой плавления 800-1000° С получают в открытых электролизерах с подиной-катодом, более (1300-1750°С) тугоплавкие в закрытых электролизерах с вертикальным катодом. Данные о температурах плавления редкоземельных металлов представлены в табл.47.
Таблица 47
Глава 10
ЭЛЕКТРОЛИЗ
Электролиз является одним из основных методов получения редких металлов, сплавов и рафинировки чернового металла.
Электролизом называется разложение электролитов постоянным электрическим током, которое сопровождается образованием новых веществ. На электродах происходят реакции окисления-восстановления: анионы на аноде отдают электроны и окисляются, а катионы восстанавливаются на катоде. Если анод растворим в электролите под действием тока, то чаще всего анионы на нем не разряжаются, а электронейтральность раствора (или расплава) поддерживается образованием катионов из материала анода. Одно из преимуществ электролиза перед химическим восстановлением заключается в том, что при этом продукты восстановления не загрязняются остатками металла-восстановителя и примесями, первоначально присутствующими в нем. Кроме того, при электролизе возможна очистка от многих примесей исходного сырья. Изменяя условия электролиза, можно получать катодный осадок с некоторыми заданными физическими свойствами (крупностью кристаллической структуры и т.п.). В промышленных масштабах осуществляют электролиз как водных растворов, так и расплавов. Однако для получения редких металлов электролиз водных растворов используют редко.
В табл. 34 приведены значения нормальных (стандартных) электродных окислительно-восстановительных потенциалов в водных растворах.
Таблица 34
Стандартные электродные окислительно-восстановительные потенциалы при 25° С в водных растворах
| Система | Eo, в | Система | Eo, в |
| Li = Li++ e- | -3,045 | Тm = Тm3+ + 3e- | -2,278 |
| К = К++ e- | -3,925 | Lu = Lu3+ + 3e- | -2,255 |
| Rb = Rb+ + e- | -2,924 | Sc = Sc3+ + 3e- | -2,077 |
| Na = Na+ + e- | -2,714 | Th = Th4+ + 4e- | -1,0899 |
| La = La3+ + 3e- | -2,522 | Be = Be2+ + 2e- | -1,847 |
| Pr = Pr3+ + 3e- | -2,462 | Hf = Hf4+ + 4e- | -1,700 |
| Nd = Nd3+ + 3e- | -2,431 | Al = Al3+ + 3e- | -1,663 |
| Sm = Sm3+ + 3e- | -3,121 | Ti = Ti2+ + 2e- | -1,630 |
| Eu = Еи2+ + 2e-- | -3,395 | Zr = Zr4+ + 4e- | -1,539 |
| Gd = Gd3+ + Зe - | -2,397 | V = V2+ + 2e- | -1,175 |
| Tb = Тb3+ + 3e- | -2,391 | Nb = Nb3+ + 3e- | -1,1 |
| Mg = Mg2+ + 2e- | -2,363 | 2Ta +5H2O = Ta2O5 + 10H+ + 10e- | -0,750 |
| Y = Y3+ + 3e- | -2,372 | Ni = Ni2+ + 2e- | -0,250 |
| Dy = Dy3++ 3e- | -2,353 | H2 = 2H2+ + 2e- | 0,000 |
| Ho = Ho3+ + 3e- | -2,319 | Cu = Cu2+ + 2e- | +0.337 |
| Ег = Ег3+ + Зe- | -2,296 |
Получение из водных растворов электроотрицательных металлов, расположенных выше водорода, невозможно, поскольку при пропускании тока через раствор выделяется водород. Лишь в некоторых случаях, когда перенапряжение водорода на поверхности выделяемого металла достаточно велико, электролиз возможен (например, выделение никеля из водных растворов). Возможен также электролиз на жидком ртутном катоде с получением амальгамы, так как перенапряжение водорода на ртути значительно.
Как видно из табл. 34, все редкие металлы, рассматриваемые в настоящем учебном пособии, электроотрицательнее водорода, вследствие чего для них приобретает особое значение процесс электролиза безводных и расплавленных сред.
При электролизе расплавов целесообразно получать металл в расплавленном состоянии, поскольку в таком виде его легче выводить из ванны (непрерывный процесс); катодный продукт после охлаждения представляет собой компактный слиток (уменьшение включений электролита). Однако большинство редких металлов (см. стр. 291) относится к числу тугоплавких. Поскольку отсутствуют доступные соли, не улетучивающиеся или не разлагающиеся при температуре выше 1500° С, тугоплавкие металлы обычно получают при температуре, более низкой, чем точка их плавления. Повышение температуры до расплав-ления металла часто связано с непреодолимыми аппаратурными затруднениями.
Металлы при электролизе в условиях tэл < tпл осаждаются на катоде в виде кристаллической губчатой массы, растущей по направлению к аноду и содержащей большое количество электролита. Опыты по получению в таких условиях гладких катод-ны.х осадков (по аналогии, например, с электролизом водных растворов меди) не дали положительных результатов. Переработка катодного продукта включает обязательную стадию отделения электролита, который часто содержит нерастворимые в воде соли. Сложность отделения электролита приводит к потерям мелкодисперсной фракции металла, и извлечение металла снижается. Иногда этих затруднений можно избежать при электролизе на жидком катоде — металле с невысокой температурой плавления. При этом образуется жидкий сплав, который либо можно использовать как таковой, либо после отгонки вспомогательного металла получить губку основного металла.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
При погружении металла в раствор электролита (или расплав) катионы могут переходить из кристаллической решетки металла в раствор (поверхность заряжается отрицательно) или из раствора в металл (поверхность приобретает положительный заряд). Возникает разность потенциалов между металлом и раствором (расплавом). При переходе катионов в раствор возникающий отрицательный заряд поверхности металла препятствует дальнейшему растворению. Поэтому величина скачка (разность потенциалов) между металлом и раствором постоянна в данных условиях.
Абсолютное значение электродного потенциала измерить невозможно, поэтому измеряют всегда разность электродных потенциалов — относительный электродный потенциал.
Если электродная реакция проходит в равновесных обратимых условиях (при токе, стремящемся к нулю), скачок потенциала между электродом и электролитом называют равновесным потенциалом. Если к погруженному в раствор металлу приложить напряжение, на бесконечно малую величину превышающее равновесный потенциал, но обратного знака, процесс, определяющий равновесный потенциал, пойдет в обратную сторону. Если первоначально металл растворялся, то произойдет выделение его на электроде - электролиз. Однако продолжительный электролиз в таких условиях осуществить не удается, так как происходящее нарушение электронейтральности раствора (выведение положительно заряженных ионов) мгновенно создаст противо-э. д. с., процесс прекратится. Для осуществления продолжительного электролиза необходимо производить одновременную разрядку отрицательных ионов раствора на втором электроде (аноде) или восполнение убыли положительных ионов. за счет растворения анода. Поскольку анод, погруженный в раствор (расплав), также обладает определенным потенциалом, то для осуществления электролиза в равновесных условиях необходимо приложить внешнее напряжение, равное сумме равновесных потенциалов анода и катода плюс бесконечно малая величина. Сумма равновесных потенциалов анода и катода называется напряжением разложения.
Равновесное значение потенциала определяется уравнением Нернста:
для катиона Ек = Еко + RTlnaMen+ / nF ; (73)
для аниона Ек = Еко - RTlnaMen- / nF ; (74)
где Е° - стандартный электродный потенциал (при активности ионов, равной 1); n - валентность; а - активность; R - газовая постоянная [8,31 дж/(моль.град)]; Т - абсолютная температура; F - число Фарадея (96520 к/г-экв).
Относительный стандартный электродный потенциал обычно приводится в таблицах для водных растворов по отношению к водородному электроду.
В случае контакта с раствором амальгамы или сплава
Е = Ео +RTln [aMen+/aMe] / nF ; (75)
где aMe - активность металла в амальгаме или сплаве; aMen+ - активность ионов металла в растворе.
Посторонние ионы (примеси других металлов, а также ионы, вводимые для создания определенной среды), даже если они электроотрицательнее основного иона и не разряжаются на катоде, в некоторых случаях существенно влияют на течение электролиза. Они влияют на коэффициенты активности вследствие образования, например, комплексных соединений, участия в процессах гидратации и дегидратации. Нейтральные ионы могут внедряться в двойной электрический слой.
Ионы металлов, более электроотрицательные, чем ион водорода, могут быть выделены из водных растворов на электроде, обеспечивающем большое перенапряжение для выделения водорода. Кроме того, можно сдвигать потенциал выделения основного металла в электроположительную область, увеличивая его активность в электролите. Регулируя (увеличивая) рН раствора, можно сдвинуть потенциал выделения водорода в электроотрицательную область.
Совместное осаждение ионов основного металла и ионов-примесей приводит к загрязнению катодного осадка. Понижение концентрации ионов-примесей в электролите резко сдвигает потенциал их выделения в электроотрицательную область, поскольку активность ионов-примесей <<1. Понижение концентрации приводит также к увеличению концентрационной поляризации. Поэтому очистка исходной соли имеет большое значение для получения осаждаемого металла высокой степени чистоты.
Напряжение разложения соли (т. е. разность обратимых потенциалов катода и анода) можно рассчитать из термохимических данных для соответствующей реакции. Например, если при электролизе происходит разложение соли МеХ3 с выделением Me и Х2, то напряжение разложения рассчитывают из термохимических данных для реакции Me+3/2 Х2 = МеХ3. Расчет сводится к вычислению энергии Гиббса DСT° реакции. При этом
ET = - DGoT / nF (76)
где n - валентность; F—число Фарадея, равное 96520 к/г-экв; DGoT - энергия Гиббса, дж/моль; Е - напряжение разложения, в.
Для приближенной оценки зависимости потенциала разложения от температуры можно пользоваться выражением
dET/dT = DSoT / nF (77)
считая энтропию DSoT не зависящей от температуры (в интервалах, где отсутствуют фазовые переходы).
Если электролизу подвергается смесь солей и происходит разложение лишь одной из них (разрядка наиболее электроположительного металла смеси), при вычислении DGoT реакции необходимо учитывать активность этой соли:
DGT = DGoT + RT In a . (78)
(если за стандартное состояние принимается расплав индивидуальной соли).
Пример расчета теоретического напряжения разложения. Рассчитать значение теоретического напряжения разложения глиноземно-криолитового расплава, если изменение энтальпии DН при 950° С для реакции 2А1+1,5О2 ® А12О3 равно -392,64 ккал, а изменение DS = -83,6 ккал/ (моль .°К)
Решение:
DG == DH -TDS = -392,64 + (273+950) . 83,6/1000 = -290,3 ккал;
-DG/nFT = Еразл = 290,3/6.23,06 = 2,1 в,
где FT =F/1000 . 4,18 = 23,06 (переход от джоулей к килокалориям).
Равновесный потенциал электрода можно определить непосредственно из эксперимента, используя обычную потенциометрическую схему, однако наличие побочных и вторичных процессов в электродной системе сильно маскирует истинное значение величины Е (к маскирующим процессам относятся пассивация электродов, образование соединений с выделяющимися продуктами и т. д.). В электрохимии водных растворов за нулевой электрод сравнения при определении Еравн исследуемого электрода принимают стандартный водородный. Кроме него в водных растворах употребляют еще несколько электродов сравнения с точно известным потенциалом - каломельный, хингидроновый, хлорсеребряный и др.
В электрохимии расплавленных солей такого универсального электрода сравнения нет. Обычно в качестве стандартного используют электрод типа Ме/МеХ, где Me - свинец, серебро, кадмий, Х - галоген. Основная трудность в этом случае при измерении Еравн в расплавах связана с необходимостью учета скачка потенциала на границе раздела двух солей - соли, в которую погружен изучаемый электрод, и расплава соли электрода сравнения.
Наряду с металлическими электродами применяют газовые электроды, обратимые к анионам. Конструктивно такие электроды представляют собой трубку из чистого графита, через которую пропускают хлор (в случае, если изучается электролиз хлоридов) или водород. Стационарный потенциал такого электрода устанавливается довольно медленно.
При установлении ряда значений напряжения выделения металлов в расплавленных солях возникают и другие трудности. Прежде всего это отсутствие стандартного состояния сравнения для разных солей: не ясно, следует сравнивать электроды при температуре плавления солей или при одинаковой температуре. Большие трудности связаны с выбором нулевого электрода.
В табл. 35 приведены электродные потенциалы некоторых металлов в их расплавленных солях.
Таблица 35
Электродные потенциалы металлов, в, в их расплавленных солях при 700° С, нулевой электрод – водородный
| электрод | анион | ||
| Cl- | Br- | I- | |
| Li/Li+ | -2,36 | -2,40 | -2,41 |
| Na/Na+ | -2,37 | -2,35 | -2,27 |
| К/К+ | -2,51 | -2,53 | -2,41 |
| Ag/Ag+ | +0,18 | -0,10 | -0,43. |
| Ве/Ве2+ | -0,90 | - | - |
| Mg/Mg2+ | -1,59 | -1,58 | -1,47 |
| Cd/Cd2+ | -0,26 | -0,46 | -0,65 |
| А1/А13+ | 0,59 | -0,57 | -0,55 |
| Bi/Bi3+ | +0,38 | +0,19 | -0,13 |
| Sb/Sb3+ | +0,53 | +0,21 | +0,03 |
Вопрос об электродных потенциалах анионов в расплавленных солях разработан слабо. Обычно сравнивают значения напряжения разложения солей с одинаковым катионом для определения положения аниона в ряду напряжений. Проще провести определение напряжения разложения соли, которое равно сумме равновесных потенциалов анода и катода. Для этого снимают зависимость тока, протекающего через ячейку электролиза, от приложенного напряжения. Прямолинейный участок зависимости I от U экстраполируется на нулевую силу тока. Соответствующее напряжение принимается равным потенциалу разложения соли в данных условиях.
В табл. 36 представлены значения напряжения разложения ряда солей.
Таблица 36
Напряжение разложения ЕТ, в, при температуре плавления некоторых галогенидов (определенное методом изучения зависимости тока от напряжения)
| катион | анион | |||
| F- | Cl- | Br- | I- | |
| Li+ | 2,55 | 3,53 | 3,22 | 2,87 |
| Na+ | 2,78 | 3,25 | 2,93 | 2,49 |
| К+' | 2,84 | 3,40 | 3,11 | 2,65 |
| Rb+ | - | 3,60 | 2,76 | 2,35 |
| Cs+ | - | 3,77 | - | 2,50 |
| Be2+ | - | 2,08 | - | - |
| Mg2+ | 2,26 | 2,59 | 2,21 | 1,75 |
| Ca2+ | 2,46 . | 2,28 | 2,82 | 2,50 |
| Sr2+ | 2,51 | 3,41 | 3,08 | 2,68 |
| Ba2+ | 2,82 | 3,44 | 3,05 | 2,36 |
| Cd2+ | - | 1,36 | 1,18 | 1,00 |
| Sn2+ | - | 1,34 | 1,09 | 0,96 |
| Pb2+ | - | 1,25 | 1,10 | 0,79 |
Разбавление индивидуальной соли другой солью с общим анионом приводит к повышению напряжения разложения, поскольку потенциал катода становится электроотрицательнее, а потенциал анода остается практически постоянным. Сдвиг потенциала катода не всегда в этом случае соответствует величине, рассчитанной по закону Нернста, так как активности ионов, как правило, не равны концентрациям.
Обычно положение металла в электрохимическом ряду сохраняется независимо от способа составления ряда и характера аниона. Наиболее электроотрицательными металлами в расплавах (как и в водных растворах) остаются щелочные металлы, затем идут щелочноземельные, элементы третьей группы и тяжелые металлы.
Однако ряд напряжений в расплавленных солях несколько меняется в зависимости от температуры, поскольку, температурные коэффициенты э. д. с. химических цепей различаются между собой. Кроме того, при переходе от одного типа анионов к другому в некоторых случаях изменяется положение металла в ряду. Так, литий в хлоридах, бромидах и иодидах более отрицателен, чем натрий, но во фторидах он положительнее натрия. Особенно большие изменения в порядке электроотрицательности металлов при изменении характера аниона характерны для тяжелых металлов (что связано со сложностью конфигурации электронных оболочек и взаимной поляризуемостью ионов).
Таким образом, электролиз расплава теоретически возможен, если к электродам приложено напряжение, лишь на бесконечно малую величину превышающее напряжение разложения соли. Однако в этом случае ток, протекающий через ячейку, также бесконечно мал, а время электролиза бесконечно велико. Для повышения скорости электролиза подается напряжение, заметно превышающее напряжение разложения. Ток при этом имеет какую-то конечную величину. При прохождении тока через электрохимическую систему происходит сдвиг потенциалов электродов от их равновесного значения: катода - в сторону более отрицательных значений, анода — в сторону более положительных значений - электрохимическая поляризация. По современным представлениям, поляризация - следствие замедленности одного или нескольких процессов, происходящих на электроде при прохождении тока. Различают концентрационную, собственно электрохимическую и химическую поляризацию. Часть общего сдвига потенциала электрода, обусловленную электрохимической поляризацией, часто называют перенапряжением.
Концентрационная поляризация обусловлена изменением концентрации потенциалообразующих веществ в приэлектродном слое. Например, разряд ионов.металла при электролизе понижает их концентрацию в слое расплава, непосредственно примыкающем к поверхности электрода. Величину концентрационной поляризации DEк можно вычислить по уравнению Нернста для концентрационных цепей:
DEк = RT/nF . lnC1/Co (79)
где С1 - концентрация потенциалообразующих ионов в приэлектродном слое; Со - концентрация в толще электролита.
При плотности тока iк на единице площади катода разряжается iк/nF г-ионов катионов, т. е. концентрация катионов в прикатодном пространстве убывает со скоростью iк/nF. Убыль катионов возмещается их переносом током. Поскольку число переноса катиона меньше единицы, поступление катионов в результате переноса током меньше убыли и, следовательно, концентрация катионов в прикатодном слое меньше, чем в толще электролита. Увеличение плотности тока приводит к такому состоянию, когда все подходящие катионы разряжаются и их концентрация у катода равна нулю. В этом случае увеличение приложенного напряжения не приведет к росту плотности тока, так как диффузия в создавшихся условиях максимальна и не в состоянии далее увеличить подвод катионов к электроду.
Постоянная и не зависимая от потенциала плотность тока называется предельной плотностью тока и рассчитывается по уравнению
iпр = DnFC / d(1-nк ) = KдС (81)
где D - коэффициент диффузии; n - валентность; F - число Фарадея; С - концентрация в объеме; d - толщина диффузного слоя; nк - число переноса катиона; Кд - константа скорости диффузии. Кд увеличивается с повышением температуры и скорости перемешивания электролита (в результате уменьшения d).
Концентрационная поляризация связана с плотностью тока уравнением
DEк = RT/nF ln (1- iк/iпр) (81)
Достижение предельного тока для данной примеси способствует очистке катодного осадка, так как дальнейшее повышение приложенного напряжения повышает скорость выделения основного металла при постоянной скорости выделения примеси.
Перемешивание электролита, повышение температуры и прочие факторы, облегчающие подачу вещества к электроду, повышают iпр и снижают концентрационную поляризацию.
Собственно электрохимической поляризацией называется смещение потенциала электрода, обусловленное только замедленностью протекания самого электрохимического процесса. Замедленность связана с тем, что электрохимическая реакция, как и всякая другая химическая реакция, требует определенной энергии активации. Наиболее высокие значения электрохимической поляризации наблюдаются при выделении газов. Возникновение перенапряжения при выделении водорода обычно связывают с замедленностью какой-либо одной или нескольких стадий этого процесса: 1) разряд иона водорода: Н+ + МеН + е- ® МеН (Me - металл, МеН - атом водорода, хемосорбированный на металле); 2) рекомбинация адсорбированных атомов: 2МеН ® Н2 + 2Me; 3) электрохимическая десорбция: Н+ + МеН + e- ® H2 + Me.
При малой плотности тока перенапряжение водорода прямо пропорционально плотности тока:
DЕ = wi
(w - константа). При большой плотности тока зависимость перенапряжения от плотности тока определяется уравнением
DЕ = a + b lg i. (82)
Повышение температуры снижает перенапряжение. Энергия активации электрохимической реакции, связанной с переходом заряда через границу электрод - раствор, изменяется с изменением потенциала электрода. Поэтому, смещая потенциал в нужную сторону (в отрицательную для катода и положительную для анода), можно снизить энергию активации электрохимического процесса и тем самым повысить его скорость до значений, обеспечивающих прохождение тока необходимой плотности.
Иногда из общего сдвига потенциала выделяют химическую поляризацию, которая определяется процессами, изменяющими химический состав поверхности электрода: покрытие поверхности анода пленкой труднорастворимых окисей, образование сплавов и интерметаллических соединений на катоде и т. п. Если концентрационную поляризацию легко определить, сравнивая, например, величины сдвига потенциала при различной скорости перемешивания электролита, то разделение электрохимической и химической поляризации часто невозможно, так как одно и то же явление может быть причиной возникновения или увеличения поляризации обоих видов. Так, образование пленок окислов приводит как к закрытию части поверхности электрода, так и к повышению энергии активации отдельных стадий процесса.
Поляризация возрастает с увеличением плотности тока, уменьшается с ростом температуры и зависит от природы электрохимической реакции, материала электрода, состава электролита и других факторов. При электролитическом осаждении металлов на катоде, как правило, большая поляризация не наблюдается.
В процессе электролитического рафинирования металла растворяется черновой анод. При этом поляризация обусловливается возрастанием концентрации ионов металла в прианодном пространстве. Пределом возрастания концентрации является насыщение и последующее выпадение осадка соли на поверхности анода, что приводит к резкому возрастанию сопротивления и падению плотности тока (солевая пассивация анода). Примеси, входящие в черновой анод, могут образовывать самостоятельные фазы. При этом потенциал анода определяется наиболее электроотрицательным компонентом смеси, более положительные компоненты не растворяются.
Частицы нерастворившихся компонентов по мере растворения электроотрицательного компонента, потеряв связь с анодом, падают на дно ванны в виде шлама.
Если электроотрицательный металл составляет меньшую часть массы анода, растворение его идет до тех пор, пока он присутствует на поверхности электрода. Затем (если поддерживать постоянную плотность тока) потенциал электрода возрастает, и начинает растворяться более благородный металл. Более сложная картина наблюдается в случае образования компонентами анода твердых растворов и химических соединений.
Электрохимическое выделение газов на аноде связано с поляризацией, причини которой установлены не во всех случаях. При выделении хлора на угольном электроде наблюдается поляризация, достигающая 0,1 в. Предполагают, что она связана с процессом адсорбции молекулярного хлора. Возможно, перенапряжение связано с замедленностью процесса образования молекул хлора из атомов.
Если выделяющийся металл взаимодействует с материалом катода, происходит деполяризация катодного процесса — сдвиг потенциала катода, в электроположительную сторону. Особенно сильна деполяризация при выделении одного жидкого металла на другом с образованием сплава. Свободная энергия реакции разряда ионов уменьшается на величину энергии взаимодействия металлов, что приводит к значительной деполяризации. Деполяризатором анодного процесса обычно служит металл, растворенный в электролите и взаимодействующий с анодными продуктами.
При электролизе возникновение поляризации нежелательно, так как приводит к дополнительному расходу электроэнергии из-за возрастания напряжения на электролитических ваннах.
Осаждение металлов на катоде в расплавленном состоянии происходит чаще всего при равновесном потенциале. Использование сложных электролитов способтсвует появлению поляризации. Осаждение твердых металлов сопровождается химической поляризацией, а также иногда поляризацией, связанной с замедленностью образования и роста кристаллов. Условия электролиза влияют на характер катодного осадка: при низкой плотности тока на поверхности кристаллов может образоваться окисная пленка - пассивация катода, увеличение поляризации. Таким образом, для практического осуществления процесса необходимо на электролизер подать напряжение, величина которого складывается из нескольких составляющих:
Uпракт = Еa – Ек + DЕa + DЕк + Еэл +Еконт, (83)
где Еа и Ек - равновесные потенциалы анода и катода; DЕa и DЕк - анодная и катодная поляризация; Еэл и Еконт - падение напряжения в электролите и контактах. В ряде случаев в Uпракт входят и другие потери: падение напряжения в диафрагме (в случае разделения катодного и анодного пространств) и т. д. При электролизе стремятся уменьшить напряжение на ячейке за счет величин поляризации и омических составляющих баланса напряжения, так как напряжение разложения (Еa – Ек) изменить нельзя.
Падение напряжения в электролите зависит от его сопротивления R=xL/s, где х -удельное сопротивление электролита, ом . см; L - расстояние между электродами; s - площадь сечения электролита, через которую проходит электрический ток, см1. В случае неравных площадей анода и катода s=Ösasк.
Величину Еэл можно уменьшить сближением электродов, введением в раствор более электропроводящих добавок, повышением температуры.
Примерный электрический баланс электролизера на 30 000 а для получения металлического натрия электролизом расплава NaCl приведен в табл. 37.
Таблица 37
Электрический баланс электролизера на 30 000 а (плотность тока на катоде 0,8-0,95, на аноде 0,9-1 а/см2)
| Составляющие баланса | Падение напряжения | |
| в | % | |
| Напряжение разложения | 3,47 | 46,5 |
| Падение напряжения в электролите | 2,00 | 25,7 |
| Падение напряжения в аноде (включая контакты) | 0,90 | 12,0 |
| Падение напряжения в катоде и токоподводах (включая контакты) | 0,48 | 7,4 |
| Остальные потери (в шинах и т. п.) | 0,62 | 8,4 |
| Всего | 7,47 | 100 |
Обеспечив подачу на электроды необходимого напряжения, можно осуществлять электролиз с заданной скоростью. Повышение приложенного напряжения приводит к увеличению тока, протекающего через электролизер, - к повышению скорости процесса.
Количество выделяемого металла определяется законом Фарадея:
q = ZIt, (84)
где q - количество вещества, получаемое на электроде, г; I - ток, а; t - время, ч; Z-электрохимический эквивалент, равный количеству граммов вещества, выделяемого при протекании через электролизер тока I а в течение 1 ч.
Электрохимические эквиваленты некоторых металлов приведены в табл. 38.
Таблица 38
Электрохимические эквиваленты некоторых металлов
| элемент | валентность | Атомная масса | Электрохимический эквивалент, г/(а.ч) | элемент | валентность | Атомная масса | Электрохимический эквивалент, г/(а.ч) |
| Li | 1 | 6,94 | 0,2589 | Pr | 3 | 140,92 | 1,7525 |
| Rb | 1 | 85,48 | 3,189 | Lu | 3 | 174,99 | 2,1765 |
| Cs | 1 | 132,91 | 4,9586 | Ti | 4 | 47,90 | 0,4467 |
| Be | 2 | 9,02 | 0,1683 | Zr | 4 | 91,22 | 0,8508 |
| Sc | 3 | 45,1 | 0,5608 | Hf | 4 | 178,6 | 2,2213 |
| Y | 3 | 138,92 | 1,7276 | Nb | 5 | 92,91 | 0,6933 |
| Ce | 3 | 140,12 | 1,7426 | Ta | 5 | 180,88 | 1,3946 |
| Ce | 4 | 140,12 | 1,3070 |
Однако не весь ток расходуется на выделение основного металла. Потери тока при электролизе расплавов в основном связаны с совместным разрядом ионов-примесей, с растворением выделяющихся на катоде металлов в расплаве. Причиной токо-потерь может служить образование соединений низшей валентности на катоде и соединений высшей валентности на аноде, испарение металлов, потери при обработке порошкообразных катодных осадков, побочные реакции продуктов электролиза с материалом электролизера, с атмосферой, а также их диффузия навстречу друг другу с последующей реакцией и образованием исходного соединения. Образование ионов низшей валентности в расплаве, содержащем многовалентные катионы, подтверждено многочисленными исследованиями. Для одновалентных ионов предполагается образование субионов типа MeMe+.
Выход по току при электролизе расплавов чаще всего понижается с повышением температуры и уменьшением плотности тока. Однако увеличение плотности тока приводит к увеличению содержания тонкодисперсной фракции, которая легко теряется при обработке катодного осадка. Добавки инертных солей повышают выход, а некоторые примеси (железа, сульфатов, влаги) снижают его.
Конструктивные особенности аппаратов для электролиза также влияют на выход по току. Уменьшение расстояния между электродами и увеличение общего количества электролита приводят к снижению выхода по току. Хорошее разделение электродных пространств друг от друга, наоборот, способствует получению высокого выхода по току.
Уменьшение растворимости металла в расплавленных солях наблюдается при введении в расплав соли более электроотрицательного металла. Иногда растворимость выделяемого металла так велика, что только введение понижающих растворимость добавок позволяет практически осуществить процесс.
Выход по току при электролизе расплавленных солей повышается также при введении небольших количеств (1-2%) поверхностно-активных веществ (например, фторидов), способствующих слиянию капель металла.
Для характеристики степени приближения условий протекания электролиза к идеальным обычно рассчитывают коэффициенты полезного использования напряжения, тока и энергии.
Коэффициент полезного использования напряжения равен отношению напряжения разложения Еразл к общему напряжению:
hн= Еразл/Uпракт
Коэффициент полезного использования тока (выход по току)
hт= b/B
где b и В - количества электричества, теоретически (по закону Фарадея) и практически расходуемые на единицу массы катодного продукта.
Коэффициент полезного использования энергии
hэ= hнhт
В электролизе расплавленных сред обычно применяют большую плотность тока - до нескольких тысяч ампер на метр квадратный. Плотность тока определяет количество продукта, получаемого с единицы электродной поверхности (производительность электролизера). Поэтому стремятся проводить процесс при максимальной плотности тока, принимая, однако, во внимание, что при этом увеличивается себестоимость продукта из-за повышения расхода электроэнергии вследствие увеличения поляризации.
При увеличении плотности тока в процессе электролиза с угольным или графитовым анодом возникает так называемый анодный эффект. Плотность тока, при которой возникает анодный эффект, называют критической. Сущность явления заключается в накоплении газа на электроде и образовании газовой пленки, отделяющей анод от жидкой среды. При повышении напряжения ток проходит через газ в результате газового ионного разряда. В газовой пленке выделяется большое количество тепла, и поверхность анода перегревается. Перегреваются и прилежащие слои электролита. Плотность тока, при которой наблюдается такое явление, зависит от природы электролита и температуры, но в среднем может быть принята равной 4-5 а/см2 для угля и 7-8 а/см2 для графита. Чаще всего это явление наступает в расплавленных фторидах, реже в хлоридах и еще реже в бромидах и иодидах. Критическая плотность тока для одного и того же электролита возрастает с температурой, с увеличением содержания окислов в расплаве.
Внешним признаком возникновения анодного эффекта служат резкое увеличение падения напряжения и уменьшение силы тока единичной ванны.
Дата: 2019-03-05, просмотров: 1095.