ЭКСТРАКЦИЯ
Экстракционные методы находят широкое применение в технологии редких металлов для очистки соединений этих металлов от примесей и для разделения близких по свойствам элементов. Применение экстракции позволяет осуществить непрерывный высокопроизводительный технологический процесс, легко поддающийся контролю и автоматизации.
ТЕРМОДИНАМИКА ЭКСТРАКЦИИ
Экстракцией называется процесс извлечения вещества из одной жидкой фазы в другую. Обычно одной фазой является водный раствор, второй - органическая жидкость, представляющая собой или чистый экстрагент, или раствор экстрагента в каком-либо инертном разбавителе. Для преодоления сил, удерживающих ионы в водной фазе, главным образом вследствие гидратации, необходима химическая связь экстрагента с извлекаемым соединением, но не слишком сильная, так как это затруднит последующую реэкстракцию.
Правило фаз
Экстракция как гетерогенное равновесие подчиняется правилу фаз Гиббса:
F = 2 + k -ф, (40)
где F - вариантность системы, т. е. число термодинамических степеней свободы; k -число компонентов (число химических веществ минус число независимых химических реакций); ф - число фаз.
Обычно в процессе экстракции температура и давление постоянны, поэтому
F'= k - ф.
Бинарная жидкостная система представляет собой насыщенный раствор одной жидкости в другой; вариантность системы равна нулю. При добавлении растворенного вещества, т. е. при увеличении числа компонентов на единицу, вариантность F становится равной единице, т. е. можно варьировать концентрацию вещества в одной из фаз. Если состав одной из фаз фиксирован, то система полностью определена: данной концентрации распределяемого вещества в водной фазе в состоянии равновесия соответствует определенная концентрация этого вещества в другой фазе.
Если в водную фазу добавить растворенное вещество до насыщения, т. е. до появления осадка, то в системе получаются 3 фазы и вариантность F становится равной нулю.
Когда в двухфазной системе растворено два компонента, то F = 4 – 2 = 2, т. е. можно фиксировать концентрацию каждого вещества в одной из фаз.
Экстракционное равновесие
Распределение вещества происходит до установления состояния равновесия. Из правила фаз следует наличие определенного отношения между концентрациями растворенного вещества в двух фазах при равновесии. Условием экстракционного равновесия является равенство химических потенциалов распределяемого вещества в обеих фазах:
mвод. = mорг.
Как известно,
m = m0 + RТ lna. (41)
где m0 - химический потенциал в стандартном состоянии; а - активность растворенного вещества. Следовательно,
mвод. = m0вод + RТ lnaвод = m0орг. + RТ lnaорг = mорг. (42)
Если за стандартное состояние принять чистое вещество, как обычно делается для неэлектролитов, то
m0вод.(чист.) + RТ lnaвод. = m0орг.(чист.) + RТ lnaорг. (43)
но
m0вод.(чист.) = m0орг.(чист).
поэтому при равновесии
aвод. = аорг, или аорг / aвод = 1
Если выразить активности через мольные доли х и абсолютные коэффициенты активности g, то
хоргgорг / хводgвод. = 1
или
хорг / хвод = gвод. / gорг
Если стандартные состояния различны, активности при равновесии будут пропорциональны. Так, для электролита удобнее за стандартное состояние принимать гипотетический раствор со свойствами бесконечно разбавленного раствора. В этом случае
mвод. = m’0вод + RТ lna’вод = m’0орг. + RТ lna’орг = mорг. (45)
аорг’ / a’вод = (1/RT) exp (Dm’0) = Kp (46)
где Dm’0 - свободная энергия перехода 1 М вещества из одного растворителя в другой в состоянии бесконечного разбавления. Из последнего уравнения следует, что активность распределяемого вещества в органической фазе пропорциональна активности в водной фазе. При фиксированной температуре Кр - величина постоянная, она называется термодинамической константой распределения вещества.
В мольных долях и концентрационных коэффициентах активности условие равновесия записывается так:
хорг / хвод = (gвод.’ / gорг’ ).Kp (47)
Наиболее важным случаем экстракционного равновесия является равновесие в системе электролит-неэлектролит, так как диэлектрическая проницаемость органической фазы, как правило, низка, а в среде с низкой диэлектрической проницаемостью диссоциация невелика:
Kдисс = exp(-Q/eKT),
где Q - энергия кулоновского взаимодействия ионов; e - диэлектрическая проницаемость; К - константа Больцмана; Т - абсолютная температура.
Пропорциональность активности извлекаемого вещества в водной и органической фазах (в формулах активность обозначена фигурными скобками) можно получить, рассматривая экстракцию как химическую реакцию, подчиняющуюся закону действующих масс:
Mn+ + nA-водн « MАn орг;
К = {MАn} орг / {Mn+} водн . {A-}n водн ; {MАn} орг / {Mn+} водн = К. {A-}n водн ;
Коэффициент распределения
Распределение вещества между фазами в состоянии равновесия характеризуется коэффициентом распределения - отношением концентраций распределяемого вещества в органической и водной фазах при равновесии:
D = Cорг./Cвод.. = [a’оргg’вод.] / [(а’вод. g’орг] = K’p . (g’вод. / g’орг ) (48)
т. е. при данной температуре он пропорционален отношению концентрационных коэффициентов активности.
Если обе фазы представляют собой растворы, идеальные по отношению к растворителю, то
Da = Cорг./Cвод.. = Kp (49)
т. е. в этом случае коэффициент распределения постоянен. Это уравнение выражает закон распределения Бертло-Нернста. На практике закон Бертло-Нернста справедлив только в случае очень разбавленных растворов (10-3 – 10-5 М), когда молекулы распределяемого вещества не взаимодействуют ни с одним из растворителей и вещество ни в одной из фаз не образует многоядерных соединений.
Экспериментально определяемый коэффициент распределения какого-либо элемента представляет собой отношение аналитически определяемых концентраций в органической и водной фазах, независимо от того, в виде каких соединений элемент в них присутствует:
Da эксп.= åCорг. i / åCвод..j
i j
ЭКСТРАГЕНТЫ
Применяемые в промышленности редких металлов экстра-генты можно разделить на три больших класса: кислые, нейтральные и основные.
Металл, находящийся в водной фазе в форме катиона или анионного комплекса, экстрагируется в органическую фазу в виде электронейтрального недиссоциированного соединения с катионом или анионом экстрагента или в виде нейтральной молекулы соли, сольватированной молекулами экстрагента.
Кислые экстрагенты
Кислые экстрагенты являются жидкими катионообменниками, содержащими способный к замещению ион водорода. К ним относятся хелатирующие экстрагенты и кислоты: фосфорорганические, карбоксильные и сульфоновые.
Хелатирующие агенты действуют как слабые кислоты:
Mm+водн + mHXорг « MXm орг + mН+водн
Обычно MXm - устойчивое мономерное соединение, которое можно выделить в твердом виде, практически нерастворимое в воде и хорошо растворимое в органических растворителях.
Хелатирующие агенты содержат также донорную группу, способную к образованию с экстрагируемым металлом бидентатного хелата. При взаимодействии иона экстрагируемого металла с хелатирующим агентом кислотный остаток экстрагента нейтрализует заряд иона металла, а оставшиеся свободными координационные места заполняются или теми же молекулами экстрагента с образованием хелата, или молекулами воды (если еще остаются свободные координационные места).
При низкой концентрации хелата органические растворы близки к идеальным, и такие системы хорошо подчиняются закону действующих масс.
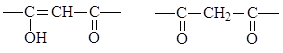 Примерами наиболее типичных хелатирующих экстрагентов являются:
Примерами наиболее типичных хелатирующих экстрагентов являются:
b -дикетоны:
енольная форма, кето-форма
способная к экстракции
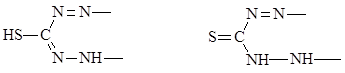 дитизон:
дитизон:
енольная форма кето-форма
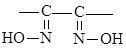
монооксимы:
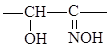
диоксимы:
Широко используемым в экстракции соединением является b-дикетон - теноилтрифторацетон (ТТА). Его енольная форма:
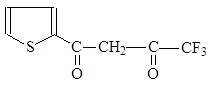 |
ТТА удобен тем, что его легко получить в чистом виде; высокая кислотность енольной формы позволяет работать с довольно кислыми растворами.
Органические кислоты (фосфорорганические, карбоксильные, сульфоновые) - обычно слабые кислоты, малорастворимые в воде. Экстрагируемые комплексы вида МХ,п дополнительно сольватированы в органической фазе, причем количество молекул экстрагента может быть различным.
Широкое распространение в экстракции получили фосфорорганические кислоты:
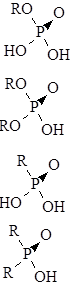
моноалкилфосфорные общего вида:
диалкилфосфорные:
моноалкилфосфоновые:
диалкилфосфоновые:
и т. д.
В органическом растворе фосфорорганические кислоты ассоциированы посредством водородных связей. Моноосновные кислоты образуют димеры, а двухосновные - полимеры различного состава.
При взаимодействии димера моноосновной кислоты с экстрагируемым ионом металла в реакцию вступает одна молекула димера, причем один ион водорода остается незамещенным.
Общее число молекул экстрагента в комплексе определяется зарядом иона:
Mm+водн + m(HX)2 орг « M(Х.НX)m орг + mН+водн
При этом образуется хелатная структура вида
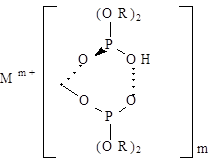
Кроме того, полученный комплекс сольватируется молекулами экстрагента до заполнения координационного числа экстрагируемого иона:
Mm+ + n/2 (HX)2 орг « MXm(n-m)НX орг + mН+водн
В зависимости от состава водной фазы в органическую фазу экстрагируются комплексы либо только с органическими анионами, либо смешанные - с неорганическими и органическими.
При высокой кислотности водного раствора, когда диссоциация органической кислоты подавлена, экстракция происходит по сольватному механизму с образованием координационной связи между атомом металла и полярным кислородным атомом экстрагента.
Одним из наиболее известных экстрагентов этого класса является ди-2-этилгексилфосфорная кислота (Д2ЭГФК):
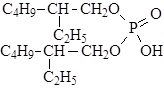
Свойства Д2ЭГФК: молекулярная масса - 322, tкип = 292°C, tвсп = 475°С, плотность 0,975 г/см3, вязкость при 25° С - 4,22 спз, nD20 = 1,4443.
Д2ЭГФК - сильный экстрагент, что в значительной степени затрудняет реэкстракцию извлекаемых соединений. При экстракции в Д2ЭГФК экстрагируемость элементов улучшается с увеличением заряда иона, а внутри каждой группы ухудшается с увеличением ионного радиуса.
Нейтральные экстрагенты
К этому классу экстрагентов относятся соединения, имеющие в своем составе атомы, способные к донорно-акцепторной связи, главным образом атомы кислорода. Кислород в экстрагенте может быть связан с атомом углерода, фосфора, серы, азота и т. д. Эти соединения экстрагируют нейтральные молекулы посредством сольватации. Неорганическая молекула сольватируется вследствие координации кислорода к центральному атому; иногда эта координация осуществляется через молекулу Н2О.
В промышленности наиболее широко используют экстрагенты двух групп, в которых атом кислорода связан с атомом фосфора или углерода. К первой группе относятся эфиры фосфорных, фосфоновых и фосфиновых кислот, а также фосфиноксиды; ко второй - простые и сложные эфиры, спирты, кетоны. Несмотря на принципиальное сходство механизма экстракции, между этими группами существуют большие различия.
Фосфорорганические соединения более полярны, чем соединения, в которых атом кислорода связан с атомом углерода. При экстракции в фосфорорганические экстрагенты вода из органической фазы обычно вытесняется, в то время как при экстракции спиртами, эфирами и кетонами вода в органической фазе необходима для образования мостиковых связей с экстрагируемым соединением. При экстракции фосфорорганическими соединениями образуется сольват с определенным числом молекул экстрагента: при экстракции в карбонилсодержащие экстрагенты образуется ряд смешанных гидратосольватов переменного состава. Большая экстрагирующая способность нейтральных фосфорорганических соединений позволяет работать с сильно разбавленными растворами экстрагентов.
Наибольшее применение из нейтральных кислородсодержащих экстрагентов получили фосфорорганические соединения общего вида: фосфаты (RO)3PO, фосфонаты (RO)2R’PO, фосфинаты (RO)(R')2PO и фосфиноксиды (R')3РО.
В ряду фосфорорганических соединений экстракционная способность изменяется в последовательности: фосфат<фосфонат<фосфинат<фосфиноксид.
Большим достоинством этих экстрагентов является низкая растворимость в водной фазе. Наименее растворимы в воде фосфаты, затем фосфонаты, фосфинаты и более всего - фосфиноксиды, причем растворимость уменьшается с увеличением длины цепи углеводородного радикала.
Из фосфорорганических соединений наиболее широко известен трибутилфосфат - ТБФ: (С4Н9О)3РО. Некоторые его свойства следующие: молекулярная масса 266, tкип = 289°C, tвсп = 146,1°С, плотность 0,973 г/см3, вязкость при 20° С – 3,32 спз, nD20 = 1,4443, при 25° С растворимость в воде 0,39 г/л, растворимость воды 64 г/л.
Молекулы ТБФ частично ассоциированы благодаря диполь-дипольному взаимодействию.
Вода растворяется в 100%-ном ТБФ с образованием моногидратов ºР®О…НОН. Образования высших гидратов экспериментально не обнаружено.
ТБФ экстрагирует минеральные кислоты, например азотную и соляную. Хлорная кислота сольватируется большим количеством молекул ТБФ, поэтому хорошо экстрагируется только при больших соотношениях ТБФ:НClO4. Серная кислота экстрагируется в ТБФ из высококонцентрированных растворов.
Экстракция металлов в ТБФ лучше всего происходит из азотнокислых растворов, поэтому экстракция нитратов изучена наиболее подробно. Например, при экстракции четырехвалентных элементов происходит реакция:
M4+ + 4NO3- « M(NO3)4.2ТБФ
При высокой кислотности возможно взаимодействие нитрата металла с сольватом азотной кислоты с образованием соединений вида M(NO3)4.2(НNO3.2ТБФ).
Основные экстрагенты
При экстракции в соединения этого класса происходит взаимодействие анионов, содержащих металл, с солями органических соединений или с катионами этих солей (катионом алкиламмония, алкиларсония или алкилфосфония).
Поскольку металл экстрагируется в составе сложных металлсодержащих комплексов, большое влияние на экстрагируемость и селективность оказывает состав водной фазы.
Из экстрагентов этого класса наибольшее значение имеют длинноцепочечные амины. Экстрагентом в данном случае является соль амина. Возможны два описания механизма экстракции солями аминов: реакцию экстракции можно представить как ионообмен или как реакцию присоединения. При ионообмене соль амина обменивает свой анион на анион, содержащий металл из водной фазы. Реакция аналогична реакциям с анионо-обменными смолами. Ее можно записать в виде:
R3Nорг + Н+ + А- « R3NH+ + А-орг;
R3NH+ + B- « R3NН+B-орг + А-;
Образовавшееся соединение может дополнительно сольватироваться солью органического основания.
С другой точки зрения, соли аминов, как и фосфорорганические соединения, можно рассматривать как экстрагенты электронодонорного типа. В солях аминов и четвертичных аммониевых оснований роль нуклеоофильного (электронодонорного) центра играет анион, входящий в состав этих солей. Способность соли металла экстрагироваться одноименной солью амина определяется способностью основного аниона - лиганда системы образовывать недиссоциированные комплексы типа МАm с катионом извлекаемого металла и, кроме того, способностью этого аниона, входящего в состав соли амина, образовывать координационную связь с атомом металла, увеличивая число присоединенных к металлу анионов-лигандов до возникновения в экстрагирующемся соединении структуры комплексного аниона [МАm+n]n-. В соли амина вида R'R''R''' NHA нуклеофильность аниона А- зависит, с одной стороны, от влияния заместителей, присоединенных к атому азота, а с другой - от природы самого аниона А-, определяемой тем, какая кислота присутствует в водной фазе, равновесной с амином.
Экстракционная способность аминов увеличивается в ряду: первичные < вторичные < третичные < четвертичные.
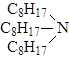 В технологии редких элементов из аминов чаще всего используется три-н-октиламин (ТОА):
В технологии редких элементов из аминов чаще всего используется три-н-октиламин (ТОА):
Молекулярная масса ТОА 353, tкип = 340°С, tвсп = 145°С, плотность 0,82 г/см3, вязкость 6,02 спз, nD20 =1,4490.
В органической фазе возможна ассоциация солей алкилами-нов в результате образования водородных связей и дипольного взаимодействия. Полимеризация солей аминов зависит от их концентрации, свойств разбавителя, длины алкильных цепей амина, температуры, а также природы соли: бисульфат и перхлорат ассоциированы в большей степени, чем нитрат и хлорид. Бисульфат триоктиламина агрегирован больше, чем сульфат. Соли три-н-октиламина в бензоле в большом интервале концентраций димеризованы.
При низких концентрациях Н2SO4 образуется сульфат амина, при более высоких - бисульфат.
При очень низких кислотностях возможна также экстракция амином по сольватному механизму.
В некоторых случаях при экстракции аминами наблюдается образование третьей фазы из-за ограниченной растворимости соли амина в разбавителе. Образование третьей фазы наиболее характерно для систем аминосульфат - алифатический углеводород. Для предотвращения образования третьей фазы к разбавителю добавляют небольшое количество длинноцепочечного спирта. Молекулы спирта взаимодействуют с ионной парой соли амина и изменяют ее диэлектрические свойства.
Синергетический эффект
Обнаружено, что определенные комбинации двух экстраген-тов при некоторых условиях лучше экстрагируют ряд металлов, чем можно было ожидать по коэффициентам распределения отдельных компонентов. Это явление называется синергетиче-ским эффектом, или синергизмом. Синергизм означает увеличение экстрагируемости металл-лигандного комплекса при добавлении в систему второй донорной молекулы:
Da эксп = DaS1 + DaS2 + DDa (50)
DDa - синергетическое увеличение.
Причиной синергетического эффекта является отчасти увеличение активности экстрагентов в смеси, но главным образом повышенная экстрагируемость комплексов смешанного состава. Синергизм проявляется не для всех металлов. Он характерен для урана, актиноидов и РЗЭ.
Известны четыре типа синергетических комбинаций экстрагентов: 1) хелатирующий агент - нейтральный сольватирующий агент, при этом между самими экстрагентами почти нет взаимодействия; 2) кислый экстрагент - нейтральный экстрагент, между ними происходит сильное взаимодействие и синергетический эффект в этом случае значительно меньше, чем в первом; 3) два нейтральных экстрагента, синергетический эффект чрезвычайно низок; 4) два хелатирующих экстрагента, при этом образуются смешанные соединения, которые могут экстрагироваться и лучше, и хуже, чем индивидуальные. Улучшение экстракции, как правило, очень незначительное.
Наконец, иногда наблюдается синёргетическая экстракция в результате совместного комплексообразования двух металлов. Например, установлено, что при экстракции небольших количеств циркония растворами 2-этилгексилфенилфосфоновой кислоты и других одноосновных фосфорсодержащих органических кислот в керосине или ароматических разбавителях в органической фазе образуются соединения циркония с экстрагентами, являющиеся более сильными экстрагентами, чем исходные фос-форорганические кислоты.
ВЫБОР ЭКСТРАГЕНТА
В технологии редких металлов в качестве экстрагентов наиболее широко применяют ТБФ, некоторые фосфинаты, фосфиноксиды, фосфорорганические кислоты и амины.
Экстрагируемость вещества зависит от экстракционной способности экстрагента и сил, удерживающих соединение в водной фазе. Наиболее объективным критерием экстракционной способности является термодинамическая константа реакции экстракции. Однако недостаточность наших знаний об активности веществ часто не позволяет вычислить значение этой константы. В таких случаях возможна оценка экстракционной способности по коэффициенту распределения экстрагируемого вещества, определяемого в строго постоянных условиях.
Экстракционная способность экстрагента определяется электронодонорными свойствами активного атома в его молекуле (в случае нейтральных фосфорорганических экстрагентов - фосфорильного кислорода, в аминах - азота). Электронодонорные свойства этих атомов объясняются наличием у них неподеленной пары электронов, благодаря чему атом кислорода или азота может образовывать координационную связь с экстрагируемым элементом.
Электронодонорные свойства зависят от строения экстрагента и ослабляются при замене алкильных радикалов (R) более электрофильными группировками (например, RO) вследствие полярного влияния групп-заместителей. При этом экстракционная способность ухудшается. Для количественной характеристики влияния строения на Электронодонорные свойства экстрагента типа R1R2R3PO или R1R2R3N предложено использовать сумму значений электроотрицательности заместителей, входящих в состав экстрагента.
Под электроотрицательностью понимают величину, количественно характеризующую способность атома, находящегося в составе устойчивой молекулы, к присоединению и отдаче электронов. Предложено характеризовать электроотрицательностью Х атома как полусумму его сродства к электрону x и ионизационного потенциала I: X = {x + I) / 2.
Такое описание электроотрицательности неоднократно подвергалось критике на основе того, что электроотрицательность атомов имеет определенное значение, в то время как реальный характер связей в различных соединениях между этими же атомами различен. Это возражение снимается развитием представлений об орбитальной электроотрицательности, зависящей от механизма образования связи.
Значение электроотрицательности рассчитывают из спектральных данных. Установлено, что электроотрицательность групп слабо зависит от длины цепочки: например, при переходе от СН3 к C6H13 электроотрицательность уменьшается от 2,07 до 2,00. При введении в молекулу экстрагента группы –ССl3 экстракционная способность снижается очень сильно.
В настоящее время не представляется возможным выполнить квантовомеханический расчет электронной плотности на активном атоме в сложной молекуле. Однако использование электроотрицательности или других характеристик полярности связи[1] и ее влияния на электронодонорные свойства экстрагента позволяет добиться хорошей корреляции зависимости экстракционной способности от строения в отдельных классах соединений.
Так, для аминов, нейтральных фосфорорганических соединений и фосфороорганических кислот предложено уравнение
lgK = A - BåX - qål, (51)
где А, В и q - константы; l - эффективная длина радикала; Х - электроотрицательность групп заместителей. BåX отражает влияние электроотрицательности групп на электронную плотность на реакционном центре (полярный или индуктивный эффект), характеризует уменьшение донорной и экстракционной способности нейтральных фосфорорганических соединений и аминов с увеличением электроотрицательности группы; для кислот - наоборот, возрастание экстракционной способности, так как для этого класса экстрагентов B<0.
Значения электроотрицательности, констант Гаммета - Тафта и Кабачника для некоторых радикалов и групп (по А. М. Розену) приведены в табл. 23.
Таблица 23
РАЗБАВИТЕЛИ
Для уменьшения вязкости и плотности органической фазы в целях улучшения расслаивания экстрагент обычно используют в виде раствора в инертном разбавителе. Под инертностью разбавителя подразумевается отсутствие экстракции в разбавитель. В экстракционной технологии учитываются экономичность и безопасность применения разбавителя. Желательно, чтобы разбавитель был дешев, не токсичен и имел высокую температуру вспышки. Чаще всего в качестве разбавителя фосфорорганических экстрагентов используют гидратированный керосин (керосиновая фракция разгонки нефти с tкип = 170-210° С после гидрирования для перевода непредельных соединений в предельные), а в случае экстракции аминами — керосин с добавкой децило-вого или октилового спирта для предотвращения образования третьей фазы.
Применение разбавителя влияет на степень идеальности органической фазы, что обусловливается тремя эффектами: 1) возрастанием энтропии раствора из-за смешения молекул различных размеров (так называемый атермический эффект, приводящий к отрицательной неидеальности, без выделения тепла); 2) наличием вандерваальсовых сил взаимодействия между молекулами, этот эффект обычно ведет к положительной неидеальности растворов; 3) химическим взаимодействием между молекулами разбавителя, экстрагента и образующихся сольватов.
Первые два эффекта обычно взаимно компенсируются, что приводит к слабой зависимости коэффициента распределения от природы разбавителя [например, при разбавлении ТБФ или диизоамилметилфосфоната (ДАМФ) керосином, ксилолом, геп-таном и т. д.].
Сильные изменения наблюдаются лишь в случае химического взаимодействия. Поскольку реакционный центр экстрагента свободен, а у сольвата блокирован, то наиболее вероятно образование химической связи разбавителя с экстрагентом, что ведет к значительному уменьшению коэффициента распределения. Например, при разбавлении ТБФ хлороформом между ними образуется водородная связь и коэффициент распределения уменьшается в 50-100 раз. К такому же результату ведет разбавление аминов спиртами.
ВЫСАЛИВАТЕЛИ
Для увеличения коэффициента распределения, а иногда и для улучшения разделения элементов в экстракционной технологии применяют высаливатели. Высаливатели сами не экстрагируются, но имеют общий ион с экстрагируемым веществом. Например, при экстракции из азотнокислых растворов в качестве высаливателей используют нитраты щелочных и щелочноземельных металлов, а также алюминия.
Высаливающее действие обусловливается в первую очередь введением одноименного иона, влияние которого ясно из рассмотрения закона действующих масс. Процесс экстрагирования с образованием сольватов в органической фазе может быть представлен в виде химической реакции
Мn+ + nА- + qS « МАn. qS
с константой равновесия
K = [МАn. qS]gс / ([Мn+] . [А-]ng±n+1 . [S]qgSq)
где gс - коэффициент активности сольвата; gS - коэффициент активности экстрагента; g± - коэффициент активности электролита.
Так как [МАn.qS]/[Мn+] = Da, то коэффициент распределения можно выразить как
Da = K . [А-]n. [S]q. g±n+1gSq/gc
При наличии высаливателя концентрация анионов определяется суммой концентрации анионов экстрагируемой соли и анионов соли высаливателя:
[A] = CM + åzCвыс (52)
где С - концентрация; z - заряд.
Повышение концентрации солей приводит к снижению диэлектрической проницаемости водной фазы, что тоже способствует образованию экстрагируемых нейтральных молекул, содержащих извлекаемый металл. Кроме того, высаливатель способствует экстракции в результате изменения химического потенциала, учитываемого в изменении коэффициента активности g±. Причиной этих изменений являются межионные и межмолекулярные взаимодействия, в частности связывание воды высаливателем, приводящее к повышению эффективной концентрации экстрагируемого иона. Действие высаливателей тем сильнее, чем сильнее гидратированы высаливаемый ион и ион самого высаликателя.
КИНЕТИКА ЭКСТРАКЦИИ
Время, требующееся для достижения равновесия, зависит от двух факторов: скорости переноса реагирующих или образующихся веществ и скорости протекающих химических реакций.
Скорость массопередачи зависит от свойств переносимых веществ, вязкости растворов, температуры, взаимной скорости движения фаз. Процесс экстракции обычно проводят при интенсивном перемешивании.
Массопередача при перемешивании осуществляется в результате конвективной диффузии распределяемого вещества в фазах и в основном молекулярной диффузии через тонкий поверхностный слой. Переход через границу раздела фаз во многих случаях сопровождается химической реакцией образования экстрагирующегося соединения.
Пограничный слой между двумя фазами - область резкого изменения концентрации распределяемого вещества. Согласно одной из наиболее ранних теорий массопередачи, теории Нернста-Льюиса-Уитмена, на границе двух несмешивающихся фаз при их относительном движении образуются две неподвижные пленки, служащие основным источником сопротивления массопередаче. Массопередача в указанных пленках осуществляется вследствие квазистационарной молекулярной диффузии, причем время установления равновесия на границе раздела фаз практически равно нулю.
При молекулярной диффузии, согласно закону Фика [см. уравнение (30)], количество диффундирующего через слой вещества пропорционально коэффициенту диффузии поверхности слоя, изменению концентрации по толщине слоя, времени и обратно пропорционально толщине слоя.
Коэффициент диффузии зависит от свойств диффундирующего вещества и среды, в. которой происходит диффузия, а также от температуры и давления.
Теоретический расчет коэффициента диффузии в жидкости сложен. Уравнения для расчета коэффициента диффузии применимы только для очень разбавленных растворов, в которых отсутствует взаимодействие растворенного вещества с растворителем. Как правило, коэффициент диффузии определяют экспериментально. В жидкости с вязкостью, близкой к вязкости воды, он имеет порядок 10-5 см2/сек.
При конвективной диффузии количество вещества, переносимое в единицу времени из фазы, отдающей распределяемое вещество, к поверхности раздела фаз (или от поверхности раздела фаз в фазу, в которую вещество переходит), пропорционально межфазному потоку j, поверхности раздела фаз s и времени t:
M = j . s . t (53)
Межфазный поток i рассчитывают через коэффициент массоотдачи b:
b = D/dэф,
где D - коэффициент диффузии; dэф - толщина диффузионного пограничного слоя (область резкого изменения концентрации вещества). В жидкостях dэф равен 0,1-0,15 доли толщины гидродинамического пограничного слоя (рис. 57).

Рис. 57. Схема изменения концентрации на границе раздела фаз: х - концентрация распределяемого вещества в водной фазе; у - в органической.
Выражение для межфазного потока получается из соотношения:
j = bx. (x - xi) = by. (yi - y) (54)
где bx и by - коэффициенты массоотдачи, а хi и уi - граничные концентрации в водной и органической фазах соответственно Условие равновесия на границе yi = Daixi, где Dai -коэффициент распределения,
x - xi = j . 1/bx; iy - y = j . 1/by; Dai . x - Dai . xi = Dai . j . 1/bx (55)
Прибавим к левой части равенства (55) выражение уi - у, а к правой 1/by, тогда
Dai . x - y = j . (1/by + Dai / bx) (56)
j = [Dai . x - y] / (1/by + Dai / bx) (57)
если Da = const, то Dax = yравн, следовательно,
j = k . (уравн – у), (58)
где k = 1 / (1/by + Dai / bx), откуда следует уравнение аддитивности фазовых сопротивлений:
1/k = 1/by + Dai / bx (59)
В случае, если экстракция сопровождается химической реакцией, например первого порядка, то в выражение для k должна войти и химическая составляющая:
1/k = 1/by + Dai / bx + 1/kхим (60)
Согласно двухпленочной теории коэффициенты массоотдачи в фазах должны быть пропорциональны коэффициентам диффузии в первой степени, что не соответствует экспериментальным данным, согласно которым они пропорциональны до D0,5-1,0.
Для устранения противоречия двухпленочной теории предложено много моделей массопередачи. По одной из них массопередача осуществляется в результате нестационарной молекулярной диффузии, многократно повторяющейся за время продвижения капли в сплошной фазе. В другой предполагается, что массопередача происходит вследствие нестационарной турбулентной диффузии. Наконец, популярна модель, согласно которой массопередача осуществляется турбулентными вихрями, при этом реализуется комбинация стационарного процесса турбулентной диффузии и нестационарного процесса молекулярной диффузии.
Однако наиболее строгое описание процесса массопередачи возможно лишь при учете реальной структуры потоков возле границы раздела фаз. Такой подход обеспечивает физико-химическая гидродинамика. Массопередача полностью определяется законом затухания турбулентных пульсации в вязком подслое.
Из выражений для конвективной и молекулярной диффузии видно, что интенсивное перемешивание ускоряет достижение равновесия вследствие увеличения поверхности раздела фаз и коэффициента массоотдачи. Однако в ряде случаев при экстракции большое значение могут приобрести сорбция и десорбция поверхностно-активных веществ (ПАВ) на границе раздела фаз. Поверхностно-активными веществами могут быть как примеси, так и сами экстрагенты и экстрагируемые вещества. Поэтому иногда истинное равновесие достигается при относительно спокойном перемешивании фаз, а при очень интенсивном - нет, так как после расслаивания поверхностно-активные вещества не успели прийти в состояние равновесия.
В технологии редких металлов обычно применяют такие экстракционные системы, в которых скорость химических реакций очень велика. Время релаксации реакции (установления равновесия) гораздо меньше времени релаксации диффузионного процесса. Такие экстракционные процессы определяют как проходящие в диффузионном режиме, так как скорость процесса определяется процессами диффузии.
Встречаются, однако, экстракционные системы с медленной химической реакцией; в этом случае скорость экстракции определяется химической реакцией и процессы протекают в кинетическом режиме. Например, скорость экстракции катионов большинства металлов фосфорорганическими кислотами велика. однако ионы алюминия, бериллия, железа экстрагируются в них медленно.
В последние годы изучению кинетики экстракционных процессов уделяется все больше внимания. Важность этих исследований заключается не только в определении путей интенсификации процесса экстракции, но в большей степени в получении информации о механизме химических реакций, сопровождающих массопередачу, а также в возможности использовать кинетические факторы для разделения методом экстракции близких по свойствам элементов.
Очистка бериллия
Методом экстракции можно получить бериллий очень высокой степени чистоты.
В качестве экстрагента используют ацетилацетон. Это b -дикетон: CH3-CO-CH2-CO-CH3. При экстракции происходит кето-енольная перегруппировка.
Кетон в енольной форме реагирует с бериллием по катионо-обменному механизму. Катион бериллия замещает водород в группе -ОН. Be(ОН)2 растворяют в смеси ацетилацетон-четыреххлористый углерод. При этом бериллий образует ацетил-ацетонат и диацетилацетонат, который растворяется в CCl4. Полученный органический раствор промывают водой и затем контактируют несколько раз с водным раствором, насыщенным ЭДТА, которая комплексует такие ионы в растворе, как Са2+, Al3+, S2+, Fe3+, Mg2+ и Cu2+. Бериллий из очищенной таким образом органической фазы реэкстрагируют азотной кислотой. Из этого раствора аммиаком осаждают гидроокись бериллия, которую затем прокаливают до ВеО. В полученной окиси бериллия, 10-6 %: Са<10, АК10, Si<10, Fe<5, Mg<5, Cu<5.
Делаются попытки очищать бериллий экстракцией из сернокислых растворов в длинноцепочечные первичные амины.
Лучше всего бериллий экстрагируется раствором 1-(3-этил-пентил)-4-этилоктиламина в CCl4. При использовании 0,3 М амина коэффициент распределения бериллия достигает 20.
Разделение РЗЭ
Экстракционное разделение лантаноидов с получением индивидуальных элементов - яркий пример огромных возможностей экстракции. Лантаноиды экстрагируются из нитратных, галогенидных, роданидных и перхлоратных растворов спиртами, простыми и сложными эфирами, кетонами, алкилфосфорными кислотами.
Целесообразно применять для разделения РЗЭ такие экстракционные методы, для которых коэффициент разделения двух рядом расположенных РЗЭ не менее 1,3.
Д2ЭГФК при концентрации 0,75 М в инертном разбавителе экстрагирует РЗЭ из 0,5 М НСl. При этом происходит реакция замещения:
3[R2POOH]2 + Ln3+ « [R2POO]-6H3Ln + 3H+
С увеличением атомного номера РЗЭ экстрагируемость его возрастает, причем коэффициент разделения двух соседних элементов равен ~2,5. Четырехвалентный церий экстрагируется значительно лучше: коэффициент распределения Се4+ при экстракции в Д2ЭГФК выше, чем коэффициент распределения Се3+ в 106 раз.
ТБФ экстрагирует РЗЭ из азотнокислых растворов при очень высокой концентрации NO3- -ионов.. Коэффициент разделения двух соседних элементов при концентрации HNO3 15,6 М составляет 1,9, если же кислотность снизить до 12М, bразд уменьшается до 1,6.
Экстрагируемость РЗЭ с возрастанием порядкового номера увеличивается, причем соблюдается линейная зависимость:
lgD = aZ - b,
где Z - атомный номер РЗЭ.
Четырехвалентный церий экстрагируется даже при низкой концентрации азотной кислоты, а при высокой кислотности рH сильно уменьшает извлечение остальных редких элементов. Установлено, что при кислотности 2 М HNO3 РЗЭ образуют с ТБФ сольват вида М(NO3)3.2ТБФ, а при 16 М HNO3 - М(NO3)3.3ТБФ и М(NO3)3.4ТБФ.
Поскольку коэффициенты разделения соседних элементов невелики, для получения индивидуальных элементов требуются десятки ступеней разделения.
Разделение ниобия и тантала
Химическое поведение ниобия и тантала характеризуется чрезвычайной склонностью к гидролизу, гидролитической полимеризации и комплексообразованию. Поэтому их водные растворы, в первую очередь азотнокислые, очень неустойчивы, что в значительной мере осложняет возможность применения экстракционных методов для разделения ниобия и тантала.
Ниобий и тантал экстрагируются нейтральными фосфорорганическими экстрагентами из 7-10 М растворов НСl (экстракция происходит в виде гидролизованных соединений), а также в виде фторметаллатных кислот из растворов HF, так как в плавиково-кислых растворах гидролиз тантала и ниобия подавляется. Из плавиковокислых растворов экстрагируется преимущественно тантал, при этом. разделение достаточно хорошее, однако экстрагируемость низка.
Экстракция из смешанных кислот позволяет получить не только хорошее разделение, но и высокие коэффициенты распределения.
Основным экстрагентом для разделения ниобия и тантала является ТБФ, который экстрагирует эти элементы из смесей HNO3-HF или H2SO4-HF. В целях предотвращения образования третьей фазы используют 100%-ный ТБФ. В органическую фазу преимущественно экстрагируется ниобий, причем его коэффициент распределения может быть очень высоким. При кислотности 6 М он достигает 80. После четырех ступеней экстракции и двух ступеней промывки 12 М HF можно извлечь 99% ниобия.
Предложен непрерывный нитрато-фторидный .процесс. Колумбит непосредственно растворяется в смеси HNO3-HF, и получается раствор, содержащий Nb 45 г/л, Та 25 г/л, 8 М HF и 3 М HNO3. Его подвергают экстракции на шести ступенях при соотношении объемов органической и водной фаз 3:1. Органическую фазу промывают раствором, содержащим 8 М HF и 3 М HNO3, при 0:В=2:1. Около 99% ниобия переходит в органический раствор, после чего реэкстрагируются 0,5 М HF при 0:В=2:3. Тантал вымывается из органической фазы раствором N2CO3, при этом достигается хорошая очистка от примесей Fe, Mn, Ti и Si. Аппаратура должна быть изготовлена из полиэтилена.
Кроме трибутилфосфата для разделения тантала и ниобия используется метилизобутилкетон или диизопропилкетон. Разделение достигает 700.
Графический метод
Для графического определения необходимо иметь данные по распределению вещества между водной и органической фазами, заданные значения исходных и конечных концентраций в фазах и отношение потоков.
Из данных по распределению строят изотерму экстракции (или кривую равновесия) y = f(x), изображающую зависимость равновесной концентрации извлекаемого вещества в органической фазе у от концентрации в водной фазе х (рис. 58). Наклон этой кривой характеризует коэффициент распределения. В идеальном случае до состояния насыщения равновесная линия должна представлять собой прямую, так как коэффициент распределения постоянен; однако в реальных системах вследствие непостоянства коэффициента распределения изотерма может считаться прямолинейной только на небольшом участке.
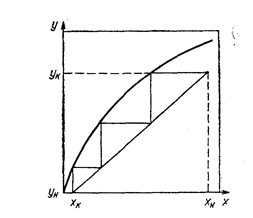
Рис. 58. Расчет числа теоретических ступеней по изотерме экстракции.
Далее строится рабочая линия, выражающая связь между концентрациями в водной и органической фазах в экстракционном аппарате на основе материального баланса. Если исходная концентрация металла в органической фазе ун, а конечная ук; исходная концентрация в водной фазе хн, а конечная хк, объем водной фазы Vводн, а органической Vорг, то в колонну входит вещество в количестве Vводнхн + Vоргун, а выходит из колонны Vводнхк + Vоргук. Эти количества должны быть равны:
Vводнхн + Vоргун = Vводнхк + Vоргук.
отсюда
ук – ун = (Vводн / Vорг) . (хн – хк)
Это уравнение прямой, проходящей через точки с координатами (хн, ук) и (хк, ун). Обычно ун = 0, хн известно, хк задается. Наклон прямой равен отношению Vорг/Vводн. Для любого сечения (рис. 59)
Vводнх + Vоргун = Vводнхк + Vоргу.
Отсюда
(у – ун) / (хк – хн) = (х – хк) / (хн – хк)
Прямая, соединяющая точки с координатами (хн, ук) и (хк, ун), наносится на график (см. рис. 58). Она называется рабочей линией. Число контактов рассчитывается графически, как показано на рисунке.
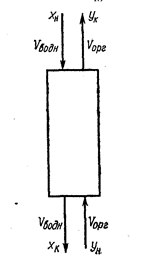
Рис. 59. Схема направления потоков в любом сечении экстракционной колонны.
Аналитический метод
Аналитический метод определения числа теоретических ступеней применяют в том случае, если коэффициент распределения можно .принять постоянным. Тогда число теоретических ступеней рассчитывают из уравнения
j = хк / хн = [E' – 1] / [E'n+1 - 1] (65)
где j - непроэкстрагированная часть вещества; Е' - коэффициент экстракции, равный DaVорг/Vводн; Da - коэффициент распределения; n - число теоретических ступеней.
Для большинства экстракционных систем этот расчет приблизительный, так как и коэффициенты распределения, и в некоторой степени потоки в процессе экстракции меняются. Если эти изменения значительны, то целесообразнее расчет проводить графически.
Обычно для получения экстракта очень высокой степени чистоты его промывают водным раствором соответствующего состава, в котором растворяется часть примесей из экстракта. Если в экстракционной системе n ступеней экстракции и т ступеней промывки, то величина j выражается уравнением
j = [(E'1 – 1).(E'2m – 1)] / [(E'1n+1 – 1).(E'2 – 1). (E'2m-1 – 1) + (E'2m-1 – 1).(E'1 – 1)] (66)
Во всех расчетах определяли число теоретических ступеней, т. е. таких ступеней, на каждой из которых достигается равновесие и фазы полностью разделяются. Но соблюдение этих условий возможно лишь на идеальной экстракционной ступени; возникает вопрос - как перейти от числа теоретических ступеней к числу реальных ступеней. В реальном экстракционном аппарате в одной его секции равновесие полностью не достигается. В принципе можно построить аппарат, в секциях которого практически полностью достигается равновесие и обеспечивается полное разделение фаз. Однако при этом пришлось бы значительно увеличить продолжительность контакта и расслаивания. Экономичнее увеличить число реальных ступеней, допуская некоторое снижение эффективности каждой из них, но зато существенно выигрывая в их объеме.
Общая эффективность (к. п. д.) экстракционного аппарата, или средняя эффективность ступени, выражается отношением числа идеальных ступеней к числу реальных ступеней экстракции:
к. п. д. = nтеор / nреальн . 100%
или
nреальн = nтеор / к. п. д. . 100%
Эффективность ступени современных смесителей-отстойников обычно не менее 70-80%. Она зависит от конструкции аппарата, определяющей тип осуществления контакта между фазами (размер капель, продолжительность контакта, скорость движения жидкости и т. д.), и коэффициента диффузии экстрагируемого компонента.
ЭКСТРАКЦИОННОЕ ОБОРУДОВАНИЕ
Для проведения процесса экстракции предложено множество конструкций аппаратов, однако только некоторые из них используют в промышленности.
Промышленные экстракционные аппараты обычно основаны на схеме противотока. Их можно разделить на две большие группы: экстракторы дифференциально-контактного типа с непрерывным изменением состава фаз и экстракторы ступенчатого типа с дискретным контактом фаз, в котором на каждой ступени происходит смешение, а затем разделение фаз.
Центробежные экстракторы
В последние годы очень широкое распространение получили экстракторы в которых для быстрого и почти полного разделения фаз используется центробежная сила. Это позволяет резко сократитить продолжительность контакта фаз (до нескольких секунд) и значительно уменьшить размеры экстрактора. Эти аппараты отличаются огромной производительностью и особенно подходящи для работы с нестойкими веществами.
В зависимости от устройства внутри центрифуги получают одну ступен"контакта пли эквивалент нескольких ступеней. Центрифуги в центробежных экстракторах располагаются горизонтально или вертикально.
Среди центробежных экстракторов дифференциального типа наиболее известен экстрактор Подбельняка (рис 63). Основной частью экстрактора является ротор, насаженный на вал и вращающийся вместе с ним со скоростью от 2000 до 5000 об/мин. Ротор представляет собой спираль из перфорированной ленты. Массообмен происходит в ее каналах (рис. 64). Тяжелая и легкая фазы подаются в аппарат насосами через полый вал. Легкая фаза подводится к периферии спирали, а тяжелая - к центру. При вращении ротора под действием центробежной силы тяжелая фаза отбрасывается к периферии, проходя через легкую. Число теоретических ступеней в одном аппарате от 3 до 10.
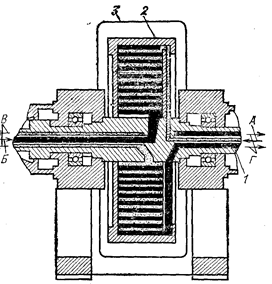
Рис. 63. Схема центробежного экстрактора Подбельняка (Кафаров В. В., 1972, рис. 245 и 246): 1 - полый вал; 2 – ротор; 3 - кожух; А - вход легкой жидкости; Б - вход тяжелой жидкости; В - выход легкой жидкости; Г - выход тяжелой жидкости.
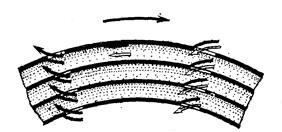
Рис. 64. Схема движения жидкости в каналах барабана центробежного, экстрактора.
Примеров вертикального центробежного экстрактора может служить экстрактор Лувеста. Этот трехступенчатый экстрактор представляет собой разновидность центробежного молочного сепаратора. В каждой ступени имеются распылительно-дисковый смеситель и центробежная осадительная камера. Экстрактор может работать с растворами, имеющими твердую взвесь.
Выбор подходящего для технологии экстрактора обычно осуществляется эмпирически, из результатов опытных испытаний.
При выборе конструкции нужно учитывать необходимое число теоретических ступеней, производительность, желательную продолжительность пребывания экстрагента, физические свойства жидкостей, в частности их плотность, вязкость, способность к эмульгированию и т. п. Сделаны попытки систематизировать имеющиеся данные по пригодности каждого типа экстрактора с точки зрения возможных требований.
[1] Например, можно использовать константу Гаммета - Тафта s* или константу Кабачника sк, представляющие собой относительную электроотрицательность. Так, константа Гаммета - Тафта связана с электроотрицательностью следующим образом: X = 2,07+0,566s*.
ЭКСТРАКЦИЯ
Экстракционные методы находят широкое применение в технологии редких металлов для очистки соединений этих металлов от примесей и для разделения близких по свойствам элементов. Применение экстракции позволяет осуществить непрерывный высокопроизводительный технологический процесс, легко поддающийся контролю и автоматизации.
ТЕРМОДИНАМИКА ЭКСТРАКЦИИ
Экстракцией называется процесс извлечения вещества из одной жидкой фазы в другую. Обычно одной фазой является водный раствор, второй - органическая жидкость, представляющая собой или чистый экстрагент, или раствор экстрагента в каком-либо инертном разбавителе. Для преодоления сил, удерживающих ионы в водной фазе, главным образом вследствие гидратации, необходима химическая связь экстрагента с извлекаемым соединением, но не слишком сильная, так как это затруднит последующую реэкстракцию.
Правило фаз
Экстракция как гетерогенное равновесие подчиняется правилу фаз Гиббса:
F = 2 + k -ф, (40)
где F - вариантность системы, т. е. число термодинамических степеней свободы; k -число компонентов (число химических веществ минус число независимых химических реакций); ф - число фаз.
Обычно в процессе экстракции температура и давление постоянны, поэтому
F'= k - ф.
Бинарная жидкостная система представляет собой насыщенный раствор одной жидкости в другой; вариантность системы равна нулю. При добавлении растворенного вещества, т. е. при увеличении числа компонентов на единицу, вариантность F становится равной единице, т. е. можно варьировать концентрацию вещества в одной из фаз. Если состав одной из фаз фиксирован, то система полностью определена: данной концентрации распределяемого вещества в водной фазе в состоянии равновесия соответствует определенная концентрация этого вещества в другой фазе.
Если в водную фазу добавить растворенное вещество до насыщения, т. е. до появления осадка, то в системе получаются 3 фазы и вариантность F становится равной нулю.
Когда в двухфазной системе растворено два компонента, то F = 4 – 2 = 2, т. е. можно фиксировать концентрацию каждого вещества в одной из фаз.
Экстракционное равновесие
Распределение вещества происходит до установления состояния равновесия. Из правила фаз следует наличие определенного отношения между концентрациями растворенного вещества в двух фазах при равновесии. Условием экстракционного равновесия является равенство химических потенциалов распределяемого вещества в обеих фазах:
mвод. = mорг.
Как известно,
m = m0 + RТ lna. (41)
где m0 - химический потенциал в стандартном состоянии; а - активность растворенного вещества. Следовательно,
mвод. = m0вод + RТ lnaвод = m0орг. + RТ lnaорг = mорг. (42)
Если за стандартное состояние принять чистое вещество, как обычно делается для неэлектролитов, то
m0вод.(чист.) + RТ lnaвод. = m0орг.(чист.) + RТ lnaорг. (43)
но
m0вод.(чист.) = m0орг.(чист).
поэтому при равновесии
aвод. = аорг, или аорг / aвод = 1
Если выразить активности через мольные доли х и абсолютные коэффициенты активности g, то
хоргgорг / хводgвод. = 1
или
хорг / хвод = gвод. / gорг
Если стандартные состояния различны, активности при равновесии будут пропорциональны. Так, для электролита удобнее за стандартное состояние принимать гипотетический раствор со свойствами бесконечно разбавленного раствора. В этом случае
mвод. = m’0вод + RТ lna’вод = m’0орг. + RТ lna’орг = mорг. (45)
аорг’ / a’вод = (1/RT) exp (Dm’0) = Kp (46)
где Dm’0 - свободная энергия перехода 1 М вещества из одного растворителя в другой в состоянии бесконечного разбавления. Из последнего уравнения следует, что активность распределяемого вещества в органической фазе пропорциональна активности в водной фазе. При фиксированной температуре Кр - величина постоянная, она называется термодинамической константой распределения вещества.
В мольных долях и концентрационных коэффициентах активности условие равновесия записывается так:
хорг / хвод = (gвод.’ / gорг’ ).Kp (47)
Наиболее важным случаем экстракционного равновесия является равновесие в системе электролит-неэлектролит, так как диэлектрическая проницаемость органической фазы, как правило, низка, а в среде с низкой диэлектрической проницаемостью диссоциация невелика:
Kдисс = exp(-Q/eKT),
где Q - энергия кулоновского взаимодействия ионов; e - диэлектрическая проницаемость; К - константа Больцмана; Т - абсолютная температура.
Пропорциональность активности извлекаемого вещества в водной и органической фазах (в формулах активность обозначена фигурными скобками) можно получить, рассматривая экстракцию как химическую реакцию, подчиняющуюся закону действующих масс:
Mn+ + nA-водн « MАn орг;
К = {MАn} орг / {Mn+} водн . {A-}n водн ; {MАn} орг / {Mn+} водн = К. {A-}n водн ;
Коэффициент распределения
Распределение вещества между фазами в состоянии равновесия характеризуется коэффициентом распределения - отношением концентраций распределяемого вещества в органической и водной фазах при равновесии:
D = Cорг./Cвод.. = [a’оргg’вод.] / [(а’вод. g’орг] = K’p . (g’вод. / g’орг ) (48)
т. е. при данной температуре он пропорционален отношению концентрационных коэффициентов активности.
Если обе фазы представляют собой растворы, идеальные по отношению к растворителю, то
Da = Cорг./Cвод.. = Kp (49)
т. е. в этом случае коэффициент распределения постоянен. Это уравнение выражает закон распределения Бертло-Нернста. На практике закон Бертло-Нернста справедлив только в случае очень разбавленных растворов (10-3 – 10-5 М), когда молекулы распределяемого вещества не взаимодействуют ни с одним из растворителей и вещество ни в одной из фаз не образует многоядерных соединений.
Экспериментально определяемый коэффициент распределения какого-либо элемента представляет собой отношение аналитически определяемых концентраций в органической и водной фазах, независимо от того, в виде каких соединений элемент в них присутствует:
Da эксп.= åCорг. i / åCвод..j
i j
ЭКСТРАГЕНТЫ
Применяемые в промышленности редких металлов экстра-генты можно разделить на три больших класса: кислые, нейтральные и основные.
Металл, находящийся в водной фазе в форме катиона или анионного комплекса, экстрагируется в органическую фазу в виде электронейтрального недиссоциированного соединения с катионом или анионом экстрагента или в виде нейтральной молекулы соли, сольватированной молекулами экстрагента.
Кислые экстрагенты
Кислые экстрагенты являются жидкими катионообменниками, содержащими способный к замещению ион водорода. К ним относятся хелатирующие экстрагенты и кислоты: фосфорорганические, карбоксильные и сульфоновые.
Хелатирующие агенты действуют как слабые кислоты:
Mm+водн + mHXорг « MXm орг + mН+водн
Обычно MXm - устойчивое мономерное соединение, которое можно выделить в твердом виде, практически нерастворимое в воде и хорошо растворимое в органических растворителях.
Хелатирующие агенты содержат также донорную группу, способную к образованию с экстрагируемым металлом бидентатного хелата. При взаимодействии иона экстрагируемого металла с хелатирующим агентом кислотный остаток экстрагента нейтрализует заряд иона металла, а оставшиеся свободными координационные места заполняются или теми же молекулами экстрагента с образованием хелата, или молекулами воды (если еще остаются свободные координационные места).
При низкой концентрации хелата органические растворы близки к идеальным, и такие системы хорошо подчиняются закону действующих масс.
Примерами наиболее типичных хелатирующих экстрагентов являются:
b -дикетоны:
енольная форма, кето-форма
способная к экстракции
дитизон:
енольная форма кето-форма
монооксимы:
диоксимы:
Широко используемым в экстракции соединением является b-дикетон - теноилтрифторацетон (ТТА). Его енольная форма:
| |
ТТА удобен тем, что его легко получить в чистом виде; высокая кислотность енольной формы позволяет работать с довольно кислыми растворами.
Органические кислоты (фосфорорганические, карбоксильные, сульфоновые) - обычно слабые кислоты, малорастворимые в воде. Экстрагируемые комплексы вида МХ,п дополнительно сольватированы в органической фазе, причем количество молекул экстрагента может быть различным.
Широкое распространение в экстракции получили фосфорорганические кислоты:
моноалкилфосфорные общего вида:
диалкилфосфорные:
моноалкилфосфоновые:
диалкилфосфоновые:
и т. д.
В органическом растворе фосфорорганические кислоты ассоциированы посредством водородных связей. Моноосновные кислоты образуют димеры, а двухосновные - полимеры различного состава.
При взаимодействии димера моноосновной кислоты с экстрагируемым ионом металла в реакцию вступает одна молекула димера, причем один ион водорода остается незамещенным.
Общее число молекул экстрагента в комплексе определяется зарядом иона:
Mm+водн + m(HX)2 орг « M(Х.НX)m орг + mН+водн
При этом образуется хелатная структура вида
Кроме того, полученный комплекс сольватируется молекулами экстрагента до заполнения координационного числа экстрагируемого иона:
Mm+ + n/2 (HX)2 орг « MXm(n-m)НX орг + mН+водн
В зависимости от состава водной фазы в органическую фазу экстрагируются комплексы либо только с органическими анионами, либо смешанные - с неорганическими и органическими.
При высокой кислотности водного раствора, когда диссоциация органической кислоты подавлена, экстракция происходит по сольватному механизму с образованием координационной связи между атомом металла и полярным кислородным атомом экстрагента.
Одним из наиболее известных экстрагентов этого класса является ди-2-этилгексилфосфорная кислота (Д2ЭГФК):
Свойства Д2ЭГФК: молекулярная масса - 322, tкип = 292°C, tвсп = 475°С, плотность 0,975 г/см3, вязкость при 25° С - 4,22 спз, nD20 = 1,4443.
Д2ЭГФК - сильный экстрагент, что в значительной степени затрудняет реэкстракцию извлекаемых соединений. При экстракции в Д2ЭГФК экстрагируемость элементов улучшается с увеличением заряда иона, а внутри каждой группы ухудшается с увеличением ионного радиуса.
Нейтральные экстрагенты
К этому классу экстрагентов относятся соединения, имеющие в своем составе атомы, способные к донорно-акцепторной связи, главным образом атомы кислорода. Кислород в экстрагенте может быть связан с атомом углерода, фосфора, серы, азота и т. д. Эти соединения экстрагируют нейтральные молекулы посредством сольватации. Неорганическая молекула сольватируется вследствие координации кислорода к центральному атому; иногда эта координация осуществляется через молекулу Н2О.
В промышленности наиболее широко используют экстрагенты двух групп, в которых атом кислорода связан с атомом фосфора или углерода. К первой группе относятся эфиры фосфорных, фосфоновых и фосфиновых кислот, а также фосфиноксиды; ко второй - простые и сложные эфиры, спирты, кетоны. Несмотря на принципиальное сходство механизма экстракции, между этими группами существуют большие различия.
Фосфорорганические соединения более полярны, чем соединения, в которых атом кислорода связан с атомом углерода. При экстракции в фосфорорганические экстрагенты вода из органической фазы обычно вытесняется, в то время как при экстракции спиртами, эфирами и кетонами вода в органической фазе необходима для образования мостиковых связей с экстрагируемым соединением. При экстракции фосфорорганическими соединениями образуется сольват с определенным числом молекул экстрагента: при экстракции в карбонилсодержащие экстрагенты образуется ряд смешанных гидратосольватов переменного состава. Большая экстрагирующая способность нейтральных фосфорорганических соединений позволяет работать с сильно разбавленными растворами экстрагентов.
Наибольшее применение из нейтральных кислородсодержащих экстрагентов получили фосфорорганические соединения общего вида: фосфаты (RO)3PO, фосфонаты (RO)2R’PO, фосфинаты (RO)(R')2PO и фосфиноксиды (R')3РО.
В ряду фосфорорганических соединений экстракционная способность изменяется в последовательности: фосфат<фосфонат<фосфинат<фосфиноксид.
Большим достоинством этих экстрагентов является низкая растворимость в водной фазе. Наименее растворимы в воде фосфаты, затем фосфонаты, фосфинаты и более всего - фосфиноксиды, причем растворимость уменьшается с увеличением длины цепи углеводородного радикала.
Из фосфорорганических соединений наиболее широко известен трибутилфосфат - ТБФ: (С4Н9О)3РО. Некоторые его свойства следующие: молекулярная масса 266, tкип = 289°C, tвсп = 146,1°С, плотность 0,973 г/см3, вязкость при 20° С – 3,32 спз, nD20 = 1,4443, при 25° С растворимость в воде 0,39 г/л, растворимость воды 64 г/л.
Молекулы ТБФ частично ассоциированы благодаря диполь-дипольному взаимодействию.
Вода растворяется в 100%-ном ТБФ с образованием моногидратов ºР®О…НОН. Образования высших гидратов экспериментально не обнаружено.
ТБФ экстрагирует минеральные кислоты, например азотную и соляную. Хлорная кислота сольватируется большим количеством молекул ТБФ, поэтому хорошо экстрагируется только при больших соотношениях ТБФ:НClO4. Серная кислота экстрагируется в ТБФ из высококонцентрированных растворов.
Экстракция металлов в ТБФ лучше всего происходит из азотнокислых растворов, поэтому экстракция нитратов изучена наиболее подробно. Например, при экстракции четырехвалентных элементов происходит реакция:
M4+ + 4NO3- « M(NO3)4.2ТБФ
При высокой кислотности возможно взаимодействие нитрата металла с сольватом азотной кислоты с образованием соединений вида M(NO3)4.2(НNO3.2ТБФ).
Основные экстрагенты
При экстракции в соединения этого класса происходит взаимодействие анионов, содержащих металл, с солями органических соединений или с катионами этих солей (катионом алкиламмония, алкиларсония или алкилфосфония).
Поскольку металл экстрагируется в составе сложных металлсодержащих комплексов, большое влияние на экстрагируемость и селективность оказывает состав водной фазы.
Из экстрагентов этого класса наибольшее значение имеют длинноцепочечные амины. Экстрагентом в данном случае является соль амина. Возможны два описания механизма экстракции солями аминов: реакцию экстракции можно представить как ионообмен или как реакцию присоединения. При ионообмене соль амина обменивает свой анион на анион, содержащий металл из водной фазы. Реакция аналогична реакциям с анионо-обменными смолами. Ее можно записать в виде:
R3Nорг + Н+ + А- « R3NH+ + А-орг;
R3NH+ + B- « R3NН+B-орг + А-;
Образовавшееся соединение может дополнительно сольватироваться солью органического основания.
С другой точки зрения, соли аминов, как и фосфорорганические соединения, можно рассматривать как экстрагенты электронодонорного типа. В солях аминов и четвертичных аммониевых оснований роль нуклеоофильного (электронодонорного) центра играет анион, входящий в состав этих солей. Способность соли металла экстрагироваться одноименной солью амина определяется способностью основного аниона - лиганда системы образовывать недиссоциированные комплексы типа МАm с катионом извлекаемого металла и, кроме того, способностью этого аниона, входящего в состав соли амина, образовывать координационную связь с атомом металла, увеличивая число присоединенных к металлу анионов-лигандов до возникновения в экстрагирующемся соединении структуры комплексного аниона [МАm+n]n-. В соли амина вида R'R''R''' NHA нуклеофильность аниона А- зависит, с одной стороны, от влияния заместителей, присоединенных к атому азота, а с другой - от природы самого аниона А-, определяемой тем, какая кислота присутствует в водной фазе, равновесной с амином.
Экстракционная способность аминов увеличивается в ряду: первичные < вторичные < третичные < четвертичные.
В технологии редких элементов из аминов чаще всего используется три-н-октиламин (ТОА):
Молекулярная масса ТОА 353, tкип = 340°С, tвсп = 145°С, плотность 0,82 г/см3, вязкость 6,02 спз, nD20 =1,4490.
В органической фазе возможна ассоциация солей алкилами-нов в результате образования водородных связей и дипольного взаимодействия. Полимеризация солей аминов зависит от их концентрации, свойств разбавителя, длины алкильных цепей амина, температуры, а также природы соли: бисульфат и перхлорат ассоциированы в большей степени, чем нитрат и хлорид. Бисульфат триоктиламина агрегирован больше, чем сульфат. Соли три-н-октиламина в бензоле в большом интервале концентраций димеризованы.
При низких концентрациях Н2SO4 образуется сульфат амина, при более высоких - бисульфат.
При очень низких кислотностях возможна также экстракция амином по сольватному механизму.
В некоторых случаях при экстракции аминами наблюдается образование третьей фазы из-за ограниченной растворимости соли амина в разбавителе. Образование третьей фазы наиболее характерно для систем аминосульфат - алифатический углеводород. Для предотвращения образования третьей фазы к разбавителю добавляют небольшое количество длинноцепочечного спирта. Молекулы спирта взаимодействуют с ионной парой соли амина и изменяют ее диэлектрические свойства.
Синергетический эффект
Обнаружено, что определенные комбинации двух экстраген-тов при некоторых условиях лучше экстрагируют ряд металлов, чем можно было ожидать по коэффициентам распределения отдельных компонентов. Это явление называется синергетиче-ским эффектом, или синергизмом. Синергизм означает увеличение экстрагируемости металл-лигандного комплекса при добавлении в систему второй донорной молекулы:
Da эксп = DaS1 + DaS2 + DDa (50)
DDa - синергетическое увеличение.
Причиной синергетического эффекта является отчасти увеличение активности экстрагентов в смеси, но главным образом повышенная экстрагируемость комплексов смешанного состава. Синергизм проявляется не для всех металлов. Он характерен для урана, актиноидов и РЗЭ.
Известны четыре типа синергетических комбинаций экстрагентов: 1) хелатирующий агент - нейтральный сольватирующий агент, при этом между самими экстрагентами почти нет взаимодействия; 2) кислый экстрагент - нейтральный экстрагент, между ними происходит сильное взаимодействие и синергетический эффект в этом случае значительно меньше, чем в первом; 3) два нейтральных экстрагента, синергетический эффект чрезвычайно низок; 4) два хелатирующих экстрагента, при этом образуются смешанные соединения, которые могут экстрагироваться и лучше, и хуже, чем индивидуальные. Улучшение экстракции, как правило, очень незначительное.
Наконец, иногда наблюдается синёргетическая экстракция в результате совместного комплексообразования двух металлов. Например, установлено, что при экстракции небольших количеств циркония растворами 2-этилгексилфенилфосфоновой кислоты и других одноосновных фосфорсодержащих органических кислот в керосине или ароматических разбавителях в органической фазе образуются соединения циркония с экстрагентами, являющиеся более сильными экстрагентами, чем исходные фос-форорганические кислоты.
ВЫБОР ЭКСТРАГЕНТА
В технологии редких металлов в качестве экстрагентов наиболее широко применяют ТБФ, некоторые фосфинаты, фосфиноксиды, фосфорорганические кислоты и амины.
Экстрагируемость вещества зависит от экстракционной способности экстрагента и сил, удерживающих соединение в водной фазе. Наиболее объективным критерием экстракционной способности является термодинамическая константа реакции экстракции. Однако недостаточность наших знаний об активности веществ часто не позволяет вычислить значение этой константы. В таких случаях возможна оценка экстракционной способности по коэффициенту распределения экстрагируемого вещества, определяемого в строго постоянных условиях.
Экстракционная способность экстрагента определяется электронодонорными свойствами активного атома в его молекуле (в случае нейтральных фосфорорганических экстрагентов - фосфорильного кислорода, в аминах - азота). Электронодонорные свойства этих атомов объясняются наличием у них неподеленной пары электронов, благодаря чему атом кислорода или азота может образовывать координационную связь с экстрагируемым элементом.
Электронодонорные свойства зависят от строения экстрагента и ослабляются при замене алкильных радикалов (R) более электрофильными группировками (например, RO) вследствие полярного влияния групп-заместителей. При этом экстракционная способность ухудшается. Для количественной характеристики влияния строения на Электронодонорные свойства экстрагента типа R1R2R3PO или R1R2R3N предложено использовать сумму значений электроотрицательности заместителей, входящих в состав экстрагента.
Под электроотрицательностью понимают величину, количественно характеризующую способность атома, находящегося в составе устойчивой молекулы, к присоединению и отдаче электронов. Предложено характеризовать электроотрицательностью Х атома как полусумму его сродства к электрону x и ионизационного потенциала I: X = {x + I) / 2.
Такое описание электроотрицательности неоднократно подвергалось критике на основе того, что электроотрицательность атомов имеет определенное значение, в то время как реальный характер связей в различных соединениях между этими же атомами различен. Это возражение снимается развитием представлений об орбитальной электроотрицательности, зависящей от механизма образования связи.
Значение электроотрицательности рассчитывают из спектральных данных. Установлено, что электроотрицательность групп слабо зависит от длины цепочки: например, при переходе от СН3 к C6H13 электроотрицательность уменьшается от 2,07 до 2,00. При введении в молекулу экстрагента группы –ССl3 экстракционная способность снижается очень сильно.
В настоящее время не представляется возможным выполнить квантовомеханический расчет электронной плотности на активном атоме в сложной молекуле. Однако использование электроотрицательности или других характеристик полярности связи[1] и ее влияния на электронодонорные свойства экстрагента позволяет добиться хорошей корреляции зависимости экстракционной способности от строения в отдельных классах соединений.
Так, для аминов, нейтральных фосфорорганических соединений и фосфороорганических кислот предложено уравнение
lgK = A - BåX - qål, (51)
где А, В и q - константы; l - эффективная длина радикала; Х - электроотрицательность групп заместителей. BåX отражает влияние электроотрицательности групп на электронную плотность на реакционном центре (полярный или индуктивный эффект), характеризует уменьшение донорной и экстракционной способности нейтральных фосфорорганических соединений и аминов с увеличением электроотрицательности группы; для кислот - наоборот, возрастание экстракционной способности, так как для этого класса экстрагентов B<0.
Значения электроотрицательности, констант Гаммета - Тафта и Кабачника для некоторых радикалов и групп (по А. М. Розену) приведены в табл. 23.
Таблица 23
Значения электроотрицательности групп-заместителей
| электроотрицательность | CH3 | C4H9 | OCH3 | ClCH2 | Cl(CH2)2 | CCl3 |
| Х | 2,07 | 2,0 | 2,9 | 2,60 | 2,34 | 2,95 |
| s* | 0 | -0,13 | 1,38 | 1,05 | 0,385 | 2,65 |
| sк | -0,95 | -1,22 | -(0,214-0,41) | -0,034 | - |
При выборе экстрагента важно учитывать его растворимость в воде и связанные с этим потери экстрагента.
Цена обычно применяемых в технологии экстрагентов лежит в пределах 100 - 2500 руб/т. Стоимость потерь может колебаться от нескольких копеек до миллиона рублей на 1 т металла. Поэтому стоимость извлекаемого металла определяет ту минимальную концентрацию элемента в водной фазе, при которой экстракция данным растворителем выгодна. Если цена экстрагента соизмерима с ценой извлекаемого вещества, то дорогие экстрагенты выгодны при потере не более 100 мг/л и концентрации извлекаемого вещества в водном растворе в несколько граммов на литр. В случае малых концентрации из-за потерь экстрагента вследствие растворимости экстракция может стать экономически невыгодной.
Существенным обстоятельством для выбора экстрагента являются также его температура вспышки, токсичность и т. д.
Экстрагенты с высокой экстракционной способностью не всегда наиболее выгодны, так как довольно большой вклад в стоимость экстракционного процесса вносит процесс регенерации экстрагента. Из экстрагентов с высокой экстракционной способностью реэкстракция затруднена.
РАЗБАВИТЕЛИ
Для уменьшения вязкости и плотности органической фазы в целях улучшения расслаивания экстрагент обычно используют в виде раствора в инертном разбавителе. Под инертностью разбавителя подразумевается отсутствие экстракции в разбавитель. В экстракционной технологии учитываются экономичность и безопасность применения разбавителя. Желательно, чтобы разбавитель был дешев, не токсичен и имел высокую температуру вспышки. Чаще всего в качестве разбавителя фосфорорганических экстрагентов используют гидратированный керосин (керосиновая фракция разгонки нефти с tкип = 170-210° С после гидрирования для перевода непредельных соединений в предельные), а в случае экстракции аминами — керосин с добавкой децило-вого или октилового спирта для предотвращения образования третьей фазы.
Применение разбавителя влияет на степень идеальности органической фазы, что обусловливается тремя эффектами: 1) возрастанием энтропии раствора из-за смешения молекул различных размеров (так называемый атермический эффект, приводящий к отрицательной неидеальности, без выделения тепла); 2) наличием вандерваальсовых сил взаимодействия между молекулами, этот эффект обычно ведет к положительной неидеальности растворов; 3) химическим взаимодействием между молекулами разбавителя, экстрагента и образующихся сольватов.
Первые два эффекта обычно взаимно компенсируются, что приводит к слабой зависимости коэффициента распределения от природы разбавителя [например, при разбавлении ТБФ или диизоамилметилфосфоната (ДАМФ) керосином, ксилолом, геп-таном и т. д.].
Сильные изменения наблюдаются лишь в случае химического взаимодействия. Поскольку реакционный центр экстрагента свободен, а у сольвата блокирован, то наиболее вероятно образование химической связи разбавителя с экстрагентом, что ведет к значительному уменьшению коэффициента распределения. Например, при разбавлении ТБФ хлороформом между ними образуется водородная связь и коэффициент распределения уменьшается в 50-100 раз. К такому же результату ведет разбавление аминов спиртами.
ВЫСАЛИВАТЕЛИ
Для увеличения коэффициента распределения, а иногда и для улучшения разделения элементов в экстракционной технологии применяют высаливатели. Высаливатели сами не экстрагируются, но имеют общий ион с экстрагируемым веществом. Например, при экстракции из азотнокислых растворов в качестве высаливателей используют нитраты щелочных и щелочноземельных металлов, а также алюминия.
Высаливающее действие обусловливается в первую очередь введением одноименного иона, влияние которого ясно из рассмотрения закона действующих масс. Процесс экстрагирования с образованием сольватов в органической фазе может быть представлен в виде химической реакции
Мn+ + nА- + qS « МАn. qS
с константой равновесия
K = [МАn. qS]gс / ([Мn+] . [А-]ng±n+1 . [S]qgSq)
где gс - коэффициент активности сольвата; gS - коэффициент активности экстрагента; g± - коэффициент активности электролита.
Так как [МАn.qS]/[Мn+] = Da, то коэффициент распределения можно выразить как
Da = K . [А-]n. [S]q. g±n+1gSq/gc
При наличии высаливателя концентрация анионов определяется суммой концентрации анионов экстрагируемой соли и анионов соли высаливателя:
[A] = CM + åzCвыс (52)
где С - концентрация; z - заряд.
Повышение концентрации солей приводит к снижению диэлектрической проницаемости водной фазы, что тоже способствует образованию экстрагируемых нейтральных молекул, содержащих извлекаемый металл. Кроме того, высаливатель способствует экстракции в результате изменения химического потенциала, учитываемого в изменении коэффициента активности g±. Причиной этих изменений являются межионные и межмолекулярные взаимодействия, в частности связывание воды высаливателем, приводящее к повышению эффективной концентрации экстрагируемого иона. Действие высаливателей тем сильнее, чем сильнее гидратированы высаливаемый ион и ион самого высаликателя.
КИНЕТИКА ЭКСТРАКЦИИ
Время, требующееся для достижения равновесия, зависит от двух факторов: скорости переноса реагирующих или образующихся веществ и скорости протекающих химических реакций.
Скорость массопередачи зависит от свойств переносимых веществ, вязкости растворов, температуры, взаимной скорости движения фаз. Процесс экстракции обычно проводят при интенсивном перемешивании.
Массопередача при перемешивании осуществляется в результате конвективной диффузии распределяемого вещества в фазах и в основном молекулярной диффузии через тонкий поверхностный слой. Переход через границу раздела фаз во многих случаях сопровождается химической реакцией образования экстрагирующегося соединения.
Пограничный слой между двумя фазами - область резкого изменения концентрации распределяемого вещества. Согласно одной из наиболее ранних теорий массопередачи, теории Нернста-Льюиса-Уитмена, на границе двух несмешивающихся фаз при их относительном движении образуются две неподвижные пленки, служащие основным источником сопротивления массопередаче. Массопередача в указанных пленках осуществляется вследствие квазистационарной молекулярной диффузии, причем время установления равновесия на границе раздела фаз практически равно нулю.
При молекулярной диффузии, согласно закону Фика [см. уравнение (30)], количество диффундирующего через слой вещества пропорционально коэффициенту диффузии поверхности слоя, изменению концентрации по толщине слоя, времени и обратно пропорционально толщине слоя.
Коэффициент диффузии зависит от свойств диффундирующего вещества и среды, в. которой происходит диффузия, а также от температуры и давления.
Теоретический расчет коэффициента диффузии в жидкости сложен. Уравнения для расчета коэффициента диффузии применимы только для очень разбавленных растворов, в которых отсутствует взаимодействие растворенного вещества с растворителем. Как правило, коэффициент диффузии определяют экспериментально. В жидкости с вязкостью, близкой к вязкости воды, он имеет порядок 10-5 см2/сек.
При конвективной диффузии количество вещества, переносимое в единицу времени из фазы, отдающей распределяемое вещество, к поверхности раздела фаз (или от поверхности раздела фаз в фазу, в которую вещество переходит), пропорционально межфазному потоку j, поверхности раздела фаз s и времени t:
M = j . s . t (53)
Межфазный поток i рассчитывают через коэффициент массоотдачи b:
b = D/dэф,
где D - коэффициент диффузии; dэф - толщина диффузионного пограничного слоя (область резкого изменения концентрации вещества). В жидкостях dэф равен 0,1-0,15 доли толщины гидродинамического пограничного слоя (рис. 57).
Рис. 57. Схема изменения концентрации на границе раздела фаз: х - концентрация распределяемого вещества в водной фазе; у - в органической.
Выражение для межфазного потока получается из соотношения:
j = bx. (x - xi) = by. (yi - y) (54)
где bx и by - коэффициенты массоотдачи, а хi и уi - граничные концентрации в водной и органической фазах соответственно Условие равновесия на границе yi = Daixi, где Dai -коэффициент распределения,
x - xi = j . 1/bx; iy - y = j . 1/by; Dai . x - Dai . xi = Dai . j . 1/bx (55)
Прибавим к левой части равенства (55) выражение уi - у, а к правой 1/by, тогда
Dai . x - y = j . (1/by + Dai / bx) (56)
j = [Dai . x - y] / (1/by + Dai / bx) (57)
если Da = const, то Dax = yравн, следовательно,
j = k . (уравн – у), (58)
где k = 1 / (1/by + Dai / bx), откуда следует уравнение аддитивности фазовых сопротивлений:
1/k = 1/by + Dai / bx (59)
В случае, если экстракция сопровождается химической реакцией, например первого порядка, то в выражение для k должна войти и химическая составляющая:
1/k = 1/by + Dai / bx + 1/kхим (60)
Согласно двухпленочной теории коэффициенты массоотдачи в фазах должны быть пропорциональны коэффициентам диффузии в первой степени, что не соответствует экспериментальным данным, согласно которым они пропорциональны до D0,5-1,0.
Для устранения противоречия двухпленочной теории предложено много моделей массопередачи. По одной из них массопередача осуществляется в результате нестационарной молекулярной диффузии, многократно повторяющейся за время продвижения капли в сплошной фазе. В другой предполагается, что массопередача происходит вследствие нестационарной турбулентной диффузии. Наконец, популярна модель, согласно которой массопередача осуществляется турбулентными вихрями, при этом реализуется комбинация стационарного процесса турбулентной диффузии и нестационарного процесса молекулярной диффузии.
Однако наиболее строгое описание процесса массопередачи возможно лишь при учете реальной структуры потоков возле границы раздела фаз. Такой подход обеспечивает физико-химическая гидродинамика. Массопередача полностью определяется законом затухания турбулентных пульсации в вязком подслое.
Из выражений для конвективной и молекулярной диффузии видно, что интенсивное перемешивание ускоряет достижение равновесия вследствие увеличения поверхности раздела фаз и коэффициента массоотдачи. Однако в ряде случаев при экстракции большое значение могут приобрести сорбция и десорбция поверхностно-активных веществ (ПАВ) на границе раздела фаз. Поверхностно-активными веществами могут быть как примеси, так и сами экстрагенты и экстрагируемые вещества. Поэтому иногда истинное равновесие достигается при относительно спокойном перемешивании фаз, а при очень интенсивном - нет, так как после расслаивания поверхностно-активные вещества не успели прийти в состояние равновесия.
В технологии редких металлов обычно применяют такие экстракционные системы, в которых скорость химических реакций очень велика. Время релаксации реакции (установления равновесия) гораздо меньше времени релаксации диффузионного процесса. Такие экстракционные процессы определяют как проходящие в диффузионном режиме, так как скорость процесса определяется процессами диффузии.
Встречаются, однако, экстракционные системы с медленной химической реакцией; в этом случае скорость экстракции определяется химической реакцией и процессы протекают в кинетическом режиме. Например, скорость экстракции катионов большинства металлов фосфорорганическими кислотами велика. однако ионы алюминия, бериллия, железа экстрагируются в них медленно.
В последние годы изучению кинетики экстракционных процессов уделяется все больше внимания. Важность этих исследований заключается не только в определении путей интенсификации процесса экстракции, но в большей степени в получении информации о механизме химических реакций, сопровождающих массопередачу, а также в возможности использовать кинетические факторы для разделения методом экстракции близких по свойствам элементов.
Дата: 2019-03-05, просмотров: 513.