ПРОФИЛАКТИЧЕСКИЕ И ЛЕЧЕБНЫЕ МЕРОПРИЯТИЯ
Агенты, имеющие целью защиту интактных клеток от повреждения (профилактические) или стимуляцию адаптивных механизмов при их альтерации (лечебные), подразделяют на немедикаментозные, медикаментозные и комбинированные.
1)Немедикаментозные агенты. Немедикаментозные средства применяют, главным образом, с целью профилактики повреждения клетки. Немедикаментозные средства повышают устойчивость клеток, органов и тканей, а также организма в целом к ряду патогенных агентов.
2)Медикаментозные средства. Лекарственные средства (ЛС) применяют в основном для активации адаптивных механизмов после воздействия патогенного агента. Большинство ЛС применяется с целью этиотропной или патогенетической терапии.
3)Комбинированные воздействия. Комбинированные воздействия дают наибольший эффект, как лечебный, так и профилактический.
ЭТИОТРОПНЫЕ, САНОГЕНЕТИЧЕСКИЕ И ПАТОГЕНЕТИЧЕСКИЕ ВОЗДЕЙСТВИЯ
Этиотропные воздействия направлены на устранение, прекращение, уменьшение силы и/или длительности действия патогенных факторов на клетки, а также на устранение условий, способствующих реализации этого действия.
Саногенетические мероприятия имеют целью активацию адаптивных механизмов (компенсации, защиты, восстановления и приспособления клеток) к изменившимся условиям.
Патогенетические воздействия направлены на разрыв звеньев механизма развития (патогенеза) патологического процесса.
№6. Клиническая и биологическая смерть, их характеристика, принципы реанимации. Биологические и социально-деонтологические аспекты реанимации. Постреанимационные состояния.
Клини́ческая смерть — обратимый этап умирания, переходный период между жизнью и смертью. На данном этапе прекращается деятельность сердца и дыхания, полностью исчезают все внешние признаки жизнедеятельности организма. При этом гипоксия (кислородное голодание) не вызывает необратимых изменений в наиболее к ней чувствительных органах и системах. Данный период терминального состояния в среднем продолжается не более 3-4 минут, максимум 5-6 минут.
Признаки клинической смерти: кома, апноэ (остановка дыхательных движений), асистолия (прекращение деятельности сердца с исчезновением биоэлектрической активности). Чем короче период между констатацией клинической смерти и началом проведения реанимационных мероприятий, тем больше шансов на жизнь у больного, поэтому диагностика и лечение проводится параллельно: 1)кома диагностируется на основании отсутствия сознания и по расширенным зрачкам, не реагирующим на свет; 2)апноэ регистрируется визуально, по отсутствию дыхательных движений грудной клетки; 3)асистолия регистрируется по отсутствию пульса на 2 сонных артериях. Перед определением пульса рекомендуется провести пострадавшему искусственную вентиляцию лёгких.
Аспекты реанимации:
Сердечно-лёгочная реанимация:
1. Базовая СЛР — это обеспечение проходимости дыхательных путей (Airway), проведение искусственной вентиляции лёгких (Breathing) и непрямого массажа сердца (Circulation). Базовая СЛР является начальным этапом оживления, когда спасатель нередко оказывается один на один с пострадавшим, и вынужден проводить реанимационные мероприятия «пустыми руками» (могут проводить непрофессиональные спасатели);
2. Специализированные реанимационные мероприятия (специализированная, или расширенная СЛР), которые должен выполнять обученный и оснащенный соответствующим оборудованием и медикаментами медицинский персонал (служба скорой медицинской помощи, врачи отделения реанимации и интенсивной терапии). Специализированная СЛР подразумевает последовательное выполнение тех же приёмов, что и при базовой СЛР, однако с использованием реанимационного оборудования, медикаментов, что и делает её существенно более эффективной:
а)аппаратный способ ИВЛ; б)ручной способ ИВЛ; в)ИВЛ «рот в рот»; г)непрямой массаж сердца; д)прямой массаж сердца; е)внутриартериальное переливание; ж)внутрисердечное введение раствора адреналина; з)дефибрилляция сердца.
Если реанимационные мероприятия не проводились или оказались безуспешными наступает биологическая смерть, которая представляет собой необратимое прекращение физиологических процессов в клетках и тканях. К ранним признакам биологической смерти относятся: 1)Отсутствие реакции глаза на раздражение (надавливание), 2) Помутнение роговицы, образование треугольников высыхания (пятен Лярше), 3)Появление симптома «кошачьего глаза»: при боковом сдавлении глазного яблока зрачок трансформируется в вертикальную веретенообразную щель, похожую на кошачий зрачок.
В дальнейшем обнаруживаются трупные пятна с локализацией в отлогих местах тела, затем возникает трупное окоченение, затем трупное расслабление, трупное разложение. Трупное окоченение и трупное разложение обычно начинаются с мышц лица, верхних конечностей. Время появления и продолжительность этих признаков зависят от исходного фона, температуры и влажности окружающей среды, причины развития необратимых перемен в организме.
Биологическая смерть субъекта не означает одномоментную биологическую смерть тканей и органов, составляющих его организм. Время до смерти тканей, составляющих тело человека, в основном определяется их способностью выживать в условиях гипоксии и аноксии. У разных тканей и органов эта способность различна. Наиболее короткое время жизни в условиях аноксии наблюдается у ткани головного мозга. Стволовые отделы и спинной мозг имеют большую сопротивляемость. Сердце сохраняет свою жизнеспособность в течение 1,5-2 часов после наступления биологической смерти. Почки, печень и некоторые другие органы сохраняют жизнеспособность до 3-4 часов. Мышечная ткань, кожа и некоторые другие ткани вполне могут быть жизнеспособными в сроки до 5-6 часов после наступления биологической смерти. Костная ткань, являясь самой инертной тканью организма человека, сохраняет свои жизненные силы до нескольких суток. С явлением переживаемости органов и тканей тела человека связана возможность трансплантации их и чем в более ранние сроки после наступления биологической смерти изымаются органы для трансплантации, чем более жизнеспособными они являются, тем больше вероятность их успешного дальнейшего функционирования в новом организме.
Периоды умирания: предагональный период, терминальная пауза, агония, клиническая смерть.
Постреанимационная болезнь - это заболевание, возникающее у пациентов после проведения успешных реанимационных мероприятий по поводу клинической смерти. Проблема становится все более актуальной из-за увеличения числа случаев успешной реанимации (оживления) больных. Главной причиной постреанимационной болезни принято считать глубокую или длительно существующую гипоксию - снижение содержания кислорода в тканях и органах, приводящее к тяжелым патологическим изменениям в них (от нарушения отдельных функций до изменения структуры тканей и органов).
СИМПТОМЫ: Проявления болезни зависят от стадии течения. В постреанимационной болезни выделяют пять основных стадий : 1- в первые 6-8 часов после успешной реанимации наступает фаза нестабильности уровня артериального давления ( артериальное давление часто падает и не обеспечивает нормальный кровоток в тканях- сохраняется синюшность слизистых оболочек, конечностей, может появиться отечность кожи ) и самостоятельного дыхания- оно угнетается и становится редким. Это приводит к ещё большему нарастанию гипоксии, она становится повреждающим элементом для системы свертывания крови с выбросом большого количества противосвертывающих факторов (определить это можно лабораторным методом). По этой причине могут наблюдаться тяжелые кровотечения. Часто в эту стадию наблюдаются внезапные остановки сердца и опасные для жизни нарушения ритма сердца (их выявляют обычно при постоянном мониторном электрокардиографическом контроле за такими пациентами). 2 стадия - относительной стабилизации основных функций организма, когда уровень гипоксии не нарастает, что можно определить лабораторным способом по уровню кислотно-основного равновесия крови и по явному улучшению самочувствия пациента. Также сохраняется высокая опасность кровотечений. Обычно всегда снижено количество выделяемой пациентом мочи. Эта фаза длится до конца первых суток послереанимационного периода. 3 фаза - повреждения органов - при сохранении гипоксии появляются признаки недостаточной работы легки (одышка в виде увеличения частоты дыханий), сердца (одышка в позе лежа, большая частота сердечных сокращений), почек (отеки, накопление в крови продуктов распада- мочевины, креатинина, высокое содержание калия), других органов (могут быть психозы, кровотечения). 4 фаза - нарушение иммунитета - развивается на 3-5 сутки и характеризуется развитием разнообразных воспалительных процессов - от пневмонии до сепсиса. Это связано с повреждением иммунного статуса высокой гипоксией. 5 фаза – исхода - зависит от течения предыдущих фаз и может быть благоприятной и неблагоприятной, что связывают со степенью поражения тканей, органов и систем организма.
№7. Понятие о реактивности организма, основные факторы, определяющие реактивность. Роль реактивности в патологии, пути и методы направленного изменения реактивности.
РЕАКТИВНОСТЬ — свойство целостного организма, обладающего нервной системой, дифференцированно (т.е. качественно и количественно определённым образом) реагировать изменением жизнедеятельности на воздействие факторов внешней и внутренней среды.
Генез реактивности. Формирование реактивности произошло по мере сочетанного усложнения следующих кардинальных характеристик живых существ: реакция — ответ организма или его части на внешнее или внутреннее воздействие; чувствительность — способность воспринимать и определять характер (качество), силу, локализацию и периодичность воздействующего на организм агента; раздражимость — свойство организма воспринимать воздействие факторов внешней и внутренней среды и отвечать на них, как правило, генерализованной, малодифференцированной реакцией, например изменением обмена веществ, формы, размеров и др.; резистентность — сопротивление, противодействие: устойчивость организма или его части к воздействию определённых факторов внешней и внутренней среды.
Категории реактивности. Реактивность определяется многими факторами и проявляется разнообразными формами изменений жизнедеятельности индивида. В связи с этим различают несколько категорий реактивности. Критериями выделения разновидностей реактивности являются основные биологические свойства организма, степень специфичности ответа организма, выраженность реакции организма на воздействие, природа агента, вызывающего ответ организма, биологическая значимость ответа организма.
Биологические свойства организма. В зависимости от основных биологических свойств организма выделены категории (виды) реактивности 1)видовая (Р, которая определяется наследственными анатомо-физиологическими особенностями представителей данного вида), 2)групповая ( Р отдельных групп особей в пределах одного вида, объединенных к-л признаком, определяющим особенности реагирования всех представителей данной группы на воздействия факторов внешней среды), 3)индивидуальная.
Степень специфичности, дифференцированности ответа организма позволяет выделить реактивность специфическую и неспецифическую.
Выраженность реакции организма на воздействие определяет реактивность нормергическую, гиперергическую (преобладают процессы возбуждения), гипергическую (пониженная реактивность) и анергию.
Природа агента. В зависимости от природы агента, вызывающего ответ организма, выделяют иммуногенную и неиммуногенную реактивность.
Биологическая значимость ответа организма определяет физиологическую и патологическую реактивность.
Таким образом, реактивность— динамичное, постоянно меняющееся свойство организма. С позиции врача важно, что это свойство можно изменять целенаправленно с целью повышения устойчивости организма к действию различных патогенных факторов.
Частные показатели состояния реактивности организма:
1)раздражимость – св-во живой кл-ки отвечать определенным образом на изменение окружающей среды4
2)возбудимость: порог возбудимости – минимальная сила раздражителя, которая способна перевести ткань из состояния покоя в состояние деятельности;
3)функциональная подвижность (лабильность);
4)хронаксия - минимальное время, требуемое для возбуждения мышечной либо нервной ткани постоянным электрическим током удвоенной пороговой силы (реобаза);
5)чувствительность – способность органов чувств приходить в состояние возбуждения при минимальной силе адекватного раздражителя.
№8. Роль наследственности в патологии. Общие закономерности патогенеза наследственных форм патологии. Механизмы их передачи.
Наследственность – свойство клеток и организмов передавать свои анатомо-физиологические признаки (особенности) потомкам. Процесс передачи этих признаков – наследование. Передача осуществляется с помощью генов – материальных единиц наследственности. От родителей потомкам передаются не признаки в готовом виде, и информация (код) о синтезе белка (фермента), детерминирующего этот признак. Гены – участки молекулы ДНК. Они состоят из кодонов. Каждый кодон представляет собой группу из 3 нуклеидов и, ≥, явл-ся нуклеотидным триплетом. Каждый кодон кодирует инфор-ию о стр-ре аминокислот и местоположение ее в белковой молекуле. Гены собираются в блоки, а последние в ДНК-нити, которые образуют хромосому. Общее число хромосом у человека в соматической кл-ке 46, в гамете – 23.
Причины наследственных болезней: Стартовое звено патогенеза наследственных заболеваний — мутации — нарушения структуры генов, хромосом или изменение их числа. В зависимости от уровня организации генетического материала (ген, хромосома, геном) говорят о мутациях генных, хромосомных и геномных.
Причинами мутаций могут быть различные факторы. Их обозначают как мутагены, а изменения, приводящие к возникновению мутаций, называют мутационным процессом. В результате мутационного процесса возникают разные виды мутаций. Изменения генетического материала разнообразны (делеции, вставки и т.д.), что позволяет подразделить мутации по механизму дефекта генетического материала (типы мутаций).
Мутагены (равно и вызываемые ими мутации) классифицируют по происхождению (источнику) на эндогенные и экзогенные, а по природе на физические, химические и биологические.
1)Экзогенные мутагены. Их большинство, к ним относятся различные и многочисленные факторы внешней среды (радиационное излучение, алкилирующие агенты, окислители, многие вирусы).
2)Эндогенные мутагены образуются в процессе жизнедеятельности организма (мутации могут возникать под влиянием свободных радикалов, продуктов липопероксидации).
1)Физические мутагены — ионизирующее излучение и температурный фактор.
2)Химические мутагены — самая многочисленная группа мутагенов. К химическим мутагенам относятся: сильные окислители или восстановители (нитраты, нитриты, активные формы кислорода); алкилирующие агенты (йодацетамид); пестициды (гербициды, фунгициды); некоторые пищевые добавки (ароматические углеводороды, цикламаты); продукты переработки нефти; органические растворители; JIC (цитостатики, содержащие ртуть средства, иммунодеп-рессанты); другие химические соединения.
3)Биологические мутагены: вирусы (например, кори, краснухи, гриппа); Аг некоторых микроорганизмов.
В результате мутаций образуется аномальный ген с измененным кодом. Реализация действия аномального гена – завершающее звено патогенеза наследственных болезней. Различают несколько путей реализации аномального гена, образовавшегося вследствие мутаций:
1-й путь реализации действия аномального гена: аномальный ген, утративший код нормальной программы синтеза структурного или функционально важного белка > прекращение синтеза иРНК > прекращение синтеза белка > нарушение ж/д > наследственная болезнь (гипоальбуминемия, гемофилия А);
2-й путь реализации действия аномального гена: аномальный ген, утративший код нормальной программы синтеза фермента > прекращение синтеза иРНК > прекращение синтеза белка-фермента > нарушение ж/д > наследственная болезнь (энзимопатическая метгемоглобинемия, гипотиреоз, альбинизм, алкаптонурия);
3-й путь реализации действия аномального гена: аномальный ген с патологическим кодом > синтез патологической иРНК > синтез патологического белка > нарушение ж/д > наследственная болезнь (серповидно - клеточная анемия).
Наследственные болезни с изменением структуры и/или числа хромосом. Одним из механизмов наследственных форм патологии является нарушение структуры или нерасхождение к-л одной или нескольких пар хромосом в процессе гаметогенеза, в связи с этим появляются патологические гаметы с недостающей или лишней хромосомой. При оплодотворении таких гамет образуются зиготы и рождаются индивиды с хромосомными болезнями (синдромы Шерешевского – Тернера (моносомия Х), Клайнфелтера (дисомия Х + моносомия Y), синдром трисомии Х-, болезнь Дауна).
№9. Основные методы изучения наследственных заболеваний. Принципы и методы выявления, профилактики и лечения.
Методы выявления: 1)клинико-генеалогический метод заключается в составлении родословной записи с последующим анализом проявления признака, характерного для конкретной наследственной болезни на протяжении возможно большего числа поколений родственников пациента.
Признаками наследственных болезней, установленных с помощью «родословных», явл-ся: 1)обнаружение болезни «по вертикали»: из поколения в поколение беспрерывно или с некоторыми перерывами; 2)менделевские соотношения между числом больных и здоровых сибсов (родные братья и сестры): 3:1, 1:1, 1:0; 3)бОльшая частота заболевания среди родственников.
2)близнецовый метод базируется на сравнительном анализе частоты определённого признака в разных группах близнецов, а также в сопоставлении с партнёрами монозиготных пар между собой и общей популяцией. Идентичность близнецов по анализируемому признаку обозначают как конкордантность, а отличие — как дискордантность. Роль наследственности и факторов среды в возникновении патологии у близнецов оценивают по специальным формулам.
3)популяционно-статистический метод заключается в составлении родословных среди большой группы населения, в пределах области или целой страны, в исследовании генетических изолятов. Изолят – группа людей, от 500 человек до нескольких тысяч, живущая изолированно от всего остального населения страны.
4)цитогенетический метод основана на микроскопическом изучении хромосом с целью выявления структурных нарушений в хромосомном наборе (кариотипирование). В качестве материала используют тканевые культуры с большим числом делящихся клеток, чаще лимфоциты периферической крови. Хромосомы на стадии метафазы изучают при помощи специальных методов окрашивания и составляют идиограммы (систематизированные кариотипы с расположением хромосом от наибольшей к наименьшей), что позволяет выявлять геномные и хромосомные мутации.
5)биохимический метод базируется на изучении биохимических показателей, отражающих сущность болезни (например, активность ферментов, наличие патологических метаболитов, концентрация компонентов ферментативной реакции).
6)молекулярная диагностика. При помощи методов ДНК-диагностики устанавливают последовательность расположения отдельных нуклеотидов, выделяют гены и их фрагменты, устанавливают их наличие в изучаемых клетках. К числу наиболее эффективных методов относятся гибридизация ДНК (блоттинг, in situ и т.д.), клонирование ДНК, полимеразная цепная реакция (ПЦР).
7)биологическое моделирование проводят для анализа возможных генетических дефектов человека с использованием в качестве объекта исследования животных (здоровых или мутантных), а также для изучения возможных мутагенных и тератогенных эффектов ЛС и других агентов, для разработки методов генной инженерии.
Принципы лечения. При лечении наследственных болезней (при соблюдении индивидуального характера помощи) применяют три основных подхода: этиотропный, патогенетический и симптоматический.
1)Этиотропный подход направлен на устранение причины заболевания. С этой целью разрабатываются, апробируются и частично могут быть применены методы коррекции генетических дефектов, называемые генной терапией. В общем виде целью генной терапии является внесение в клеточный геном поражённых органов нормально экспрессируемого «здорового» гена, выполняющего функцию мутантного («больного») гена. Цель патогенетической терапии — разрыв звеньев патогенеза. Для достижения этой цели применяют несколько методов.
2)Заместительная терапия — введение в организм дефицитного вещества (не синтезирующегося в связи с аномалией гена, который контролирует продукцию данного вещества; например, инсулина при СД, соответствующих ферментов при гликогенозах и агликогенозах, антигемофильного глобулина человека при гемофилии). Коррекция метаболизма путём ограничения попадания в организм веществ, метаболически не усваивающихся; выведения из организма метаболитов, накапливающихся в нём в избытке; регуляции активности ферментов. Хирургическая коррекция дефектов.
3)Симптоматическая терапия направлена на устранение симптомов, усугубляющих состояние пациента.
Методы профилактики. Медико-генетическое консультирование является основным видом профилактики врождённой и наследственной патологии. Задача консультирования: сформулировать прогноз для потомства, течения заболевания, качества жизни и здоровья.
Пренатальная диагностика осуществляется в I и II триместрах беременности (в периоды, когда возможно прерывание беременности при обнаружении патологии плода).
Методы диагностики: Ультразвуковое исследование, Биохимическое исследование сыворотки крови матери, Фетоскопия, Цитогенетические, биохимические и молекулярно-генетические методы.
Преклиническая диагностика — скрининг с целью ранней диагностика наследственных болезней обмена веществ у новорождённых.
Диспансеризация семей с наследственной патологией проводится с целью предупреждения рождения больного ребёнка или зачатия аномального плода (первичная профилактика).
Контроль мутагенной опасности факторов окружающей среды реализуется путём предотвращения образования, снижения содержания, длительности и/или силы действия на организм химических, физических и биологических мутагенных агентов. Достигается комплексом организационных и гигиенических мер на производстве, в учреждениях и быту (например, возведением очистных сооружений; применением спецодежды, очисткой воздуха, воды и продуктов питания; использованием средств противорадиационной защиты).
№10. Ионизирующая радиация как патогенный фактор. Механизмы его патогенного действия и последствия.
Ионизирующее излучение может действовать на организм как из внешних, так и из внутренних источников облучения. Человек подвергается действию ИИ в производственных условиях, работая с рентгеновской аппаратурой, на ядерных реакторах и ускорителях заряженных частиц. Источником внутр облучения могут быть радиоактивные вещ-ва, поступающие в организм с пищей, водой, через кожные покровы. По своей природе все ИИ делятся на электромагнитные (рентгеновское излучение и гама-лучи, сопровождающие радиоактивный распад) и корпускулярные (заряженные частицы: ядра гелия – α-лучи, электроны – β-лучи, протоны, π-мезотоны; нейроны, не несущие электрического заряда). Повреждающее действие различных видов ионизир радиации зависит от величины плотности ионизации в тканях и их проникающей способности. Чем короче путь прохождения фотонов и частиц в тканях, тем больше вызванная ими плотность ионизации и сильнее повреждающее действие. Для сравнительной количественной оценки биологического действия различных видов излучения определяют их относительную биологическую эффективность (наиб ОБЭ - α-излучения, протоны). Единицей измерения дозы является грей(Гр). Облучение может быть однократным, дробным и длительным.
Механизм патогенного действия: биологическое действие ионизирующей радиации выражается в развитии местных лучевых р-ий (ожоги и катаракты) и особого генерализованного процесса – лучевой болезни. В процессе повреждающего действия выделяют: а)первичное действие ИИ; б)влияние радиации на кл-ки; в)действие радиации на целый организм:
а) первичное действие ИИ на живую ткань проявляется ионизацией, возбуждением атомов и молекул и образованием при этом свободных радикалов – пусковой механизм биологич-го действия излучений. Непрямое действие ИР связано с радиационн-химич-ми изменениями стр-ры ДНК, ферментов, белков, вызываемыми продуктами радиолиза воды или растворенных в ней вещ-в. При окислении ненасыщ жир к-т и фенолов обр-ся липидные и хиноновые первичные радиотоксины, угнетающие синтез НК, подавляющие активность разл ферментов, повыш-ие проницаемость мембран и изменяющие диффузные процессы в кл-ке ≥ нарушение процессов обмена, функ и структ повреждения кл-ок, органов и систем орг-ма.
б) действие ИР на клетки: ИИ вызывает разл р-ии кл-ок – от временной задержки размножения до их гибели. По радиочувствительности кл-ок ткани можно расположить: лимфоидные органы, костный мозг, семенники, яички, слиз обол ЖКТ, эпителий кожи. В основе радиационного поражения кл-ок лежат нарушения ультраструктуры органелл и связанные с этим изменения обмена вещ-в. Малые дозы ИИ вызывают обратимые, не летальные изменения кл-ки (появляются сразу или через неск минут после облучения и стечением времени исчезают): ингибирование нуклеинового обмена, изменение проницаемости мембран, возникновение липкости хромосом, образование зерен и глыбок в ядерном вещ-ве, задержка митозов. При больших дозах облучения в кл-ах наступают летальные изменения, приводящие к их гибели до вступления в митоз либо в момент митотического деления; гибель клеток ведет к опустошению тканей, нарушению из стр-ры и ф-ии.
в) действие ИО на организм может быть местным(лучевые ожоги, некрозы, катаракты) и общим(лучевая болезнь). Местное действие ИР чаще проявляется в виде лучевых ожогов. Мягкое рентгеновское и β-излучение, проникающие в ткани на незначительную глубину, вызывают ожоги кожи, высокоэнергетическое тормозное γ-излучение и нейроны, обладающие большей проникающей способностью, могут поражать и глубоко лежащие ткани. Течение лучевых ожогов характер-ся развитием последовательно сменяющихся периодов( ранняя лучевая реакция, скрытое, острое воспаление, восстановление), длительность и выраженность проявлений которых зависит от тяжести поражения (Iстепени – 8-12 Гр – легкие; IIстепени – 12-20 Гр – средней тяжести; IIIстепени – более 20 Гр – тяжелые). При облучении дозами более 20 Гр погибает не только кожа, но и подкожная клетчатка, фасции, мышцы и даже кости. У больных развивается лихорадка, высокий лейкоцитоз, тяжелый болевой синдром. Лучевая болезнь: при внешнем равномерном облучении организма в зависимости от дозы ИР возникают поражения от едва уловимых р-ий со стороны отдельных систем до острых форм лучевой болезни. При облучении в дозах 1-10 Гр развивается типичная форма острой лучевой болезни(костнмозговая), при которой наиболее четко проявляются основные патогенетич закономерности клинического формирования ее отдельных периодов, имеет место преимущественное поражение костного мозга. В диапазоне доз 10-20 Гр возникает кишечная; при 20—80 Гр - токсемическая; выше 80Гр – церебральная форма ЛБ. При воздействии ионизирующих излучений возникают характерные патоморфологические изменения, которые схематически можно разделить на четыре группы: 1) дистрофические изменения в различных органах и тканях; 2) геморрагические проявления; 3) опустошение кроветворных органов; 4) инфекционные осложнения.
Отдаленные последствия действия радиации: 1)неопухолевые формы: сокращение продолжительности жизни, гипопластические состояния в кроветворной ткани,слиз обол органов ЖКТ, дыхательных путей, в коже; склеротические процессы, дисгормональные состояния; 2)опухолевые формы: развитие опухолей в критическких органах, радиационные лейкозы.
№11. Местные и общие реакции на действие ионизирующего излучения. Дозы излучения.
Местное действие ИР чаще проявляется в виде лучевых ожогов. Мягкое рентгеновское и β-излучение, проникающие в ткани на незначительную глубину, вызывают ожоги кожи, высокоэнергетическое тормозное γ-излучение и нейроны, обладающие большей проникающей способностью, могут поражать и глубоко лежащие ткани. Течение лучевых ожогов характеризуется развитием последовательно сменяющихся периодов (ранняя лучевая реакция, скрытое, острое воспаление, восстановление), длительность и выраженность проявлений которых зависит от тяжести поражения (I степени – 8-12 Гр – легкие; II степени – 12-20 Гр – средней тяжести; III степени – более 20 Гр – тяжелые). При облучении дозами более 20 Гр погибает не только кожа, но и подкожная клетчатка, фасции, мышцы и даже кости. У больных развивается лихорадка, высокий лейкоцитоз, тяжелый болевой синдром. Лучевая болезнь (общие реакции): при внешнем равномерном облучении организма в зависимости от дозы ИР возникают поражения от едва уловимых реакций со стороны отдельных систем до острых форм лучевой болезни. При облучении в дозах 1-10 Гр развивается типичная форма острой лучевой болезни( костномозговая), при которой наиболее четко проявляются основные патогенетические закономерности клинического формирования ее отдельных периодов, имеет место преимущественное поражение костного мозга. В диапазоне доз 10-20 Гр возникает кишечная; при 20—80 Гр - токсемическая; выше 80Гр – церебральная форма ЛБ. При воздействии ионизирующих излучений возникают характерные патоморфологические изменения, которые схематически можно разделить на четыре группы: 1) дистрофические изменения в различных органах и тканях; 2) геморрагические проявления; 3) опустошение кроветворных органов; 4) инфекционные осложнения.
№12. Острая лучевая болезнь. Клинические формы. Периоды развития костномозговой формы острой лучевой болезни. Картина крови на разных стадиях болезни.
При внешнем равномерном облучении организма в зависимости от дозы ИР возникают поражения от едва уловимых реакций со стороны отдельных систем до острых форм лучевой болезни. При облучении в дозах 1-10 Гр развивается типичная форма острой лучевой болезни (костномозговая), при которой наиболее четко проявляются основные патогенетические закономерности клинического формирования ее отдельных периодов, имеет место преимущественное поражение костного мозга. В диапазоне доз 10-20 Гр возникает кишечная; при 20—80 Гр - токсемическая; выше 80Гр – церебральная форма ЛБ. Типичная форма острой лучевой болезни по тяжести поражения, определяемая поглощенной дозой излучения, классифицируется на 4 группы: 1 – легкой степени (1-2 Гр); 2 – средней степени (2-4 Гр); 3 – тяжелой степени (4-6 Гр); 4 – крайне тяжелой степени (свыше 6 Гр). В ее течении выделяют 4 фазы: 1) первичной острой реакции; 2) мнимого клинического благополучия (скрытая фаза); 3) разгара болезни; 4) восстановления.
1)Период первичной острой реакции: Первые симптомы общей первичной реакции следуют либо непосредственно после облучения, либо через несколько часов. У пораженных внезапно появляются тошнота и рвота, общая слабость, головные боли, головокружение, общее возбуждение, а иногда угнетение и апатия, вялость, сонливость. Часто больные ощущают жажду и сухость во рту, в некоторых случаях возникают периодические боли в подложечной области и внизу живота, сердцебиения, боли в области сердца. В тяжелых случаях рвота принимает характер многократной или неукротимой, появляется жидкий стул или понос, тенезмы, парез желудка или кишечника, общая слабость достигает степени адинамии, развивается выраженное психомоторное возбуждение. При объективном исследовании в этот период выявляются гиперемия кожи, гипергидроз, лабильность вазомоторов, тремор пальцев рук, тахикардия, повышение в первые часы артериального давления, а затем снижение его. В крайне тяжелых случаях обнаруживаются иктеричность склер, патологические рефлексы, менингеальные симптомы и повышение температуры тела, может развиться сердечно-сосудистая недостаточность.
При исследовании крови (картина крови) определяются нейтрофильный лейкоцитоз со сдвигом влево или без него, относительная и абсолютная лимфопения, наклонность к ретикулоцитозу. В костном мозге уменьшено содержание миелокариоцитов, эритробластов и число митозов, повышен цитолиз.
Первичная реакция продолжается от нескольких часов до трех-четырех суток, в дальнейшем ее проявления уменьшаются или исчезают, наступает второй, скрытый период заболевания – период относительного клинического благополучия. Продолжительность фазы первичной острой реакции 1-3 дня.
2)Период мнимого благополучия: он характеризуется включением в патологический процесс защитных механизмов организма. Самочувствие больных становится удовлетворительным, проходят клинически видимые признаки болезни. Однако в скрытом периоде, несмотря на улучшение самочувствия пациентов, выявляются признаки прогрессирующих нарушений функционального состояния систем крови, нервной и эндокринной систем, дистонические и обменные расстройства. У пострадавших обнаруживаются признаки астенизации и вегето-сосудистой неустойчивости, они жалуются на повышенную утомляемость, потливость, периодические головные боли, неустойчивость настроения, расстройство сна, снижение аппетита. Характерны лабильность пульса с тенденцией к тахикардии, наклонность к гипотонии, при более тяжелых поражениях – ослабление тонов сердца.
Картина крови: наблюдавшийся в начальном периоде лейкоцитоз сменяется лейкопенией с нейтропенией, лимфопенией, уменьшается число ретикулоцитов, а с конца первой недели появляется тромбоцитопения. Наблюдаются качественные изменения клеток: гиперсегментация ядер нейтрофилов, полиморфизм ядер лимфоцитов, токсическая зернистость в протоплазме нейтрофилов. При биохимических исследованиях крови определяется диспротеинемия с тенденцией к снижению содержания альбуминов, повышению альфа-глобулинов, появляется С-реактивный белок. Продолжительность скрытого периода различна. В крайне тяжелых случаях он может отсутствовать, а при более легких – достигает трех-четырех недель.
3)Период разгара начинается с ухудшения самочувствия и общего состояния. В дальнейшем выявляются признаки прогрессирующего нарушения кроветворения и обмена веществ, присоединяются инфекционные осложнения, в тяжелых случаях развивается картина сепсиса, возникают кровоточивость, гектическая лихорадка с ознобами и проливными потами, появляются кровоточивость и кровоизлияния в кожу, слизистые оболочки, желудочно-кишечный тракт, мозг, сердце, легкие. В результате нарушений обмена веществ, электролитного гомеостаза, обезвоживания и диспепсических расстройств резко снижается масса тела.
Пульс учащен, границы сердца сдвигаются на периферию, тоны сердца становятся глухими, а над верхушкой выслушивается систолический шум. Часто присоединяются бронхит и пневмония. В тяжелых случаях на фоне диспепсических расстройств и резкого снижения аппетита возникает язвенный или язвенно-некротический стоматит, глоссит, тонзиллит, энтероколит. Из-за резкой болезненности слизистых десен и полости рта и болей при глотании пациент не может принимать пищу, а упорные поносы приводят к быстрому истощению.
Проявления кровоточивости раньше всего обнаруживаются на слизистых полости рта, в последующем кровоизлияния образуются на коже паховых областей, на внутренних поверхностях бедер, голеней, предплечий, в нижнем треугольнике живота; в тяжелых случаях присоединяются носовые и кишечные кровотечения, кровоизлияния в сетчатку, гематурия.
Волосы начинают выпадать на голове, лобке, затем на подбородке, в подмышечных впадинах и на туловище.
При неврологическом исследовании отмечаются выраженная заторможенность больных, астенизация, иногда симптомы раздражения мозговых оболочек, анизорефлексия, понижение сухожильных и периостальных рефлексов, мышечная гипотония, снижение брюшных рефлексов.
На глазном дне – застойные явления с мелкими кровоизлияниями. На электрокардиограмме регистрируются признаки ухудшения функционального состояния миокарда: снижение вольтажа, уширение желудочкового комплекса, удлинение систолического показателя, уплощение зубцов Т и Р, смещение интервала S-Т.
Картина крови: нарушения кроветворения достигают наибольшей выраженности в соответствии с дозой поражения. В тяжелых случаях возникает упорная пангемоцитопения. Число лейкоцитов снижается до 0,2-0,05×109/л, содержание тромбоцитов – до 5-10×109/л. Прогрессирует анемия. Количество миелокариоцитов в костном мозге уменьшается до 3-5×109/л, сам костный мозг представляется гипо- или апластичным, а его клеточный состав представлен ретикулярными, эндотелиальными и плазматическими клетками, единичными резко измененными лимфоцитами и сегментоядерными нейтрофилами, отсутствуют ретикулоциты.
На высоте заболевания нарушается гемостаз: удлиненяется время свертывания и длительность кровотечения, нарушается ретракция кровяного сгустка, замедляется время рекальцифнкации, тромбиновое время, снижаются толерантность крови к гепарину и потребление протромбина, степень тромботеста и активность фибринстабилизирующего фактора, усиливается фибринолитическая и снижается антифбринолитическая активность крови.
Общее содержание белка в сыворотке крови уменьшено, снижено количество альбуминов и увеличено содержание альфа-1- и, в особенности, альфа-2-глобулинов.
При бактериологическом исследовании в период выраженных клинических проявлений, активизации инфекции из крови и костного мозга высевается разнообразная флора (чаще всего кишечная палочка, стафилококк и стрептококк).
Период разгара продолжается от двух до четырех недель.
4)Период восстановления. Он начинается с появления признаков оживления кроветворения. В периферической крови обнаруживаются вначале единичные промиелоциты, миелоциты, моноциты и моноцитоиды, ретикулоциты, в дальнейшем быстро в течение нескольких дней нарастает число лейкоцитов, тромбоцитов и ретикулоцитов. В костном мозге выявляется картина бурной регенерации с большим числом бластных форм, митозов, прогрессирующим увеличением общего числа миелокариоцитов. Выход из агранулоцитоза наступает тем раньше, чем раньше он начался, т.е. чем выше доза. Однако при дозах облучения более 6 Гр выход из агранулоцитоза будет задерживаться из-за резчайшего уменьшения запасов стволовых клеток. Период агранулоцитоза заканчивается окончательным восстановлением уровня лейкоцитов и тромбоцитов. Выход из агранулоцитоза обычно бывает быстрым – в течение 1-3 дней, нередко ему на 1-2 дня предшествует подъем тромбоцитов. К моменту выхода из агранулоцитоза возрастает и уровень ретикулоцитов, нередко существенно превышая нормальный (репаративный ретикулоцитоз), однако уровень эритроцитов в это время самый минимальный (через 1-1,5 месяца).
Одновременно с началом регенерации кроветворения с увеличением числа нейтрофилов происходит критическое падение температуры тела, улучшение общего самочувствия, исчезновение признаков кровоточивости. Однако восстановление измененных функций идет медленно, в течение длительного времени остаются астенизация, вегетативно-сосудистая дистония, лабильность гематологических показателей, нарушение функционального состояния гипоталамо-гипофизарно-надпочечниковой системы, ряд трофических и обменных расстройств. Нарушены нервные процессы: торможения, возбуждения, их уравновешенность и подвижность, регистрируются явления раздражительной слабости, наличие фазовых состояний коры больших полушарий. В некоторых случаях наблюдаются вегетативно-сосудистые пароксизмы, развернутый диэнцефальный синдром, вестибулярные нарушения.
Период восстановления в тяжелых случаях продолжается от нескольких месяцев до одного года, в дальнейшем иногда на протяжении многих лет выявляются остаточные явления или отдаленные соматические и генетические последствия.
К отдаленным последствиям относятся ряд неврологических синдромов (астеновегетативный, диэнцефальный, радиационный энцефаломиелоз), сокращение продолжительности жизни, развитие катаракты, понижение плодовитости, возникновение лейкозов и новообразований. Генетические последствия обычно не выявляются у самого пострадавшего, а обнаруживаются при статистическом изучении его потомства. Они выражаются в повышении в потомстве облученных родителей числа новорожденных с пороками развития, в увеличении детской смертности и числа выкидышей и мертворожденных, изменении соотношения количества рождаемых мальчиков и девочек. Степень генетических и соматических последствий увеличивается по мере возрастания дозы радиационного поражения. Выраженность симптомов в том или ином периоде и продолжительность отдельных периодов определяются степенью тяжести лучевой болезни.
№13. Острая лучевая болезнь. Кишечная, токсемическая и мозговая формы острой лучевой болезни.
При внешнем равномерном облучении организма в зависимости от дозы ИР возникают поражения от едва уловимых реакций со стороны отдельных систем до острых форм лучевой болезни. При облучении в дозах 1-10 Гр развивается типичная форма острой лучевой болезни (костномозговая), при которой наиболее четко проявляются основные патогенетические закономерности клинического формирования ее отдельных периодов, имеет место преимущественное поражение костного мозга. В диапазоне доз 10-20 Гр возникает кишечная; при 20—80 Гр - токсемическая; выше 80Гр – церебральная форма ЛБ.
1)Кишечная форма острой лучевой болезни возникает при дозе облучения 10-20 Гр. В клинической картине заболевания на первый план выступают симптомы поражения желудочно-кишечного тракта (кишечная форма); характерна тяжелая, длительная (до 4-5 суток) первичная реакция в виде неукротимой рвоты, головной боли, тахикардии, гипотонии, наблюдаются эритема кожи, иктеричностъ склер, субфебрильное повышение температуры тела. Состояние пострадавшего резко ухудшается со второй недели заболевания: появляются симптомы острейшего энтероколита, высокая лихорадка, признаки обезвоживания организма, изменения слизистых рта и глотки, инфекционные осложнения. Развиваются геморрагия и глубокая лейкопения с полным отсутствием лимфоцитов в периферической крови, а также картина сепсиса. Смерть, причиной которой являются дегидратация организма, сопровождающаяся потерей электролитов и белка, развитие необратимого шока, связанного с действием токсических веществ микробного и тканевого происхождения, наступает на второй-третьей неделе от момента поражения.
2)Токсемическая форма острой лучевой болезни наступает при дозах облучения в пределах 20-80 Гр. В клинической картине ведущими являются прогрессирующие нарушения функционального состояния нервной системы, на фоне которых развиваются признаки острой сердечно-сосудистой недостаточности. Сразу после облучения появляются неукротимая рвота, понос, нарастают симптомы общей интоксикации, возникают гипертермия, тахикардия, возможен коллапс. Сознание обычно сохранено, наблюдаются адинамия, сильнейшие головные боли, головокружение; на фоне сосудистой недостаточности развивается олиго-, анурия, азотемия. В основе таких нарушений лежит интоксикация продуктами тканевого распада – токсемическая форма лучевой болезни. Смерть наступает на 4-8-е сутки.
3)Церебральная форма острой лучевой болезни возникает при облучении в дозах выше 80 Гр. Смерть наступает через 1-3 дня после облучения, а при действии очень больших доз (150-200 Гр) смертельный исход может иметь место даже в ходе самого облучения («смерть под лучом») или через несколько минут-часов после воздействия, а также при локальном облучении головы дозами 100-300 Гр.
Эта форма лучевого поражения характеризуется развитием судорожно-паралитического синдрома, дезориентацией, атаксией, психомоторным возбуждением, затемнением и потерей сознания, расстройством дыхания и кардиоваскулярными нарушениями, изнуряющей рвотой и поносами с примесью крови в рвотных массах и кале, коллапсом, олиго-, анурией, клоническими и тоническими судорогами. Смертельный исход наступает на протяжении первых трех суток в связи с нарушениями крово- и лимфообращения в ЦНС, сосудистого тонуса и падением кровяного давления и расстройством терморегуляции.
Причиной смерти при церебральной форме острой лучевой болезни являются тяжелые и необратимые нарушения ЦНС, характеризующиеся значительными структурными изменениями, гибелью клеток коры головного мозга и нейронов ядер гипоталамуса. В поражении нервной системы главную роль играют непосредственное повреждающее действие радиации на ткань, а также первичные радиотоксины в виде Н2О2 и других веществ, образующихся за счет окисления ненасыщенных жирных кислот и фенолов.
Отдаленные последствия действия радиации: 1)неопухолевые формы: сокращение продолжительности жизни, гипопластические состояния в кроветворной ткани,слиз обол органов ЖКТ, дыхательных путей, в коже; склеротические процессы, дисгормональные состояния; 2)опухолевые формы: развитие опухолей в критическких органах, радиационные лейкозы.
№14. Хроническая лучевая болезнь. Этиология, патогенез, проявления, исходы. Картина крови.
ХЛБ возникает при длительном облучении организма в малых дозах, но превышающих допустимые дозах. Выделяют два основных варианта болезни: 1)обусловленной внешним общим или местным облучением, 2)в результате поступления в организм равномерно и неравномерно распределяющихся радиоактивных нуклидов.
Заболевание отличается постепенным развитием и длительным волнообразным течением, сроки возникновения и характер изменений при этом определяются интенсивностью и суммарной дозой облучения. Начальный период заболевания характеризуется развитием нестойкой лейкопении, признаками астенизации, вегетативно-сосудистой неустойчивостью. Развёрнутому периоду заболевания свойственная недостаточность физиологической регенерации наиболее радиочувствительных тканей в сочетании с функциональными изменениями в деятельности нервной и ссс. Период восстановления характеризуется сглаживанием деструктивных и отчетливым преобладанием репаративных процессов в наиболее радиопоражаемых тканях. По тяжести ХЛБ, обусловленную общим облучением, делят на: легкой (I), средней (II) и тяжелой (III) степени.
1) ХЛБ I степени(легкая) характеризуется нерезко выраженными нервно-регуляторными нарушениями в деятельности различных органов и систем, умеренной нестойкой лейкопенией и тромбоцитопенией.
2) При ХЛБ II степени (средней) присоединяются функциональные нарушения нервной, с-с и пищеварительной систем. Прогрессирует лейкопения и лимфопения, количество тромбоцитов уменьшено; в костном мозге – явление гипоплазии кроветворения.
3) При ХЛБ III степени (тяжелой) развиваются анемия, явления выраженной гипоплазии кроветворения, атрофические процессы в слизистой ЖКТ, присоединяются инфекционно-септические осложнения, геморрагический синдром и нарушения кровообращения. Крайне тяжелые формы встречаются редко, при этом у больных развиваются поносы и кахексия.
Клиническую картину ХЛБ, обусловленной внутренним облучением, формирует поражение одного или нескольких критических органов, в которых депонируются поступившие в организм радиоактивные нуклиды.
Отдаленные последствия действия радиации: 1)неопухолевые формы: сокращение продолжительности жизни, гипопластические состояния в кроветворной ткани,слиз обол органов ЖКТ, дыхательных путей, в коже; склеротические процессы, дисгормональные состояния; 2)опухолевые формы: развитие опухолей в критическких органах, радиационные лейкозы.
№15. Гипертермические состояния. Причины, стадии и общие механизмы развития. Отличие лихорадки от экзогенной гипертермии.
ГИПЕРТЕРМИЯ — типовая форма расстройства теплового обмена, возникающая в результате действия высокой температуры окружающей среды и/или нарушения процессов теплоотдачи организма; характеризуется нарушением (срывом) механизмов теплорегуляции, проявляется повышением температуры тела выше нормы.
Причинами гипертермии являются: 1) высокая температура окружающей среды, 2) агенты, препятствующие реализации механизмов теплоотдачи организма, 3) разобщители процессов окисления и фосфорилирования в митохондриях.
Стадии гипертермии. Гипертермия, как правило, процесс стадийный. При действии гипертермического фактора в организме включается триада экстренных адаптивных реакций: 1) поведенческая («уход» от действия теплового фактора), 2) интенсификация процессов теплоотдачи и снижение активности теп-лопродукции, 3) стресс-реакция.
В ходе развития гипертермии условно выделяют две основные стадии: 1)компенсации (адаптации); 2)декомпенсации (деадаптации) механизмов терморегуляции организма. Иногда выделяют финальную стадию гипертермии — гипертермическую кому.
Механизм развития гипертермии включает комплекс адаптивных и патогенных реакций организма. На начальной стадии доминируют первые, на последующих (если компенсаторные и защитные реакции оказались недостаточными) — преобладают процессы повреждения. На каждой из стадий гипертермии в организме развиваются характерные метаболические, физико-химические, структурные и функциональные изменения.
Стадия компенсации характеризуется активацией экстренных механизмов адаптации организма к перегреванию. Эти механизмы направлены на увеличение теплоотдачи и снижение теплопродукции. В результате температура тела хотя и повышается, но остаётся в пределах верхней границы нормального диапазона. Проявления гипертермии в значительной мере определяются температурой окружающей среды.
Стадия декомпенсации характеризуется срывом и неэффективностью как центральных, так и местных механизмов терморегуляции, что и приводит к нарушению температурного гомеостаза организма.
ТЕПЛОВОЙ УДАР. Тепловой удар —своеобразная форма гипертермии. Это своеобразие заключается в остроте развития гипертермии с быстрым достижением опасных для жизни значений температуры тела (ректальной) 42-43 °С. Тепловой удар — следствие быстрого истощения и срыва приспособительных процессов, характерных для стадии компенсации гипертермии.
Причины: Действие теплового фактора высокой интенсивности; низкая эффективность механизмов адаптации организма к повышенной температуре внешней среды.
Патогенез: Перегревание организма после кратковременной (иногда клинически не определяемой) стадии компенсации быстро приводит к срыву механизмов терморегуляции и интенсивному нарастанию температуры тела. Последняя имеет тенденцию приближаться к температуре внешней среды. Следовательно, тепловой удар — гипертермия с непродолжительной стадией компенсации, быстро переходящая в стадию декомпенсации.
Последствия: Смерть пациентов при тепловом ударе является результатом острой прогрессирующей интоксикации, сердечной недостаточности, остановки дыхания.
СОЛНЕЧНЫЙ УДАР. Солнечный удар, являясь одной из форм гипертермических состояний, имеет ряд отличий от гипертермии как по причине, так и по механизмам развития.
Причина. Причиной солнечного удара является прямое воздействие энергии солнечного излучения на организм. Наибольшее патогенное действие, наряду с другими, оказывает инфракрасная часть солнечной радиации, т.е. радиационное тепло. Последнее, в отличие от конвекционного и кондукционного тепла, одновременно прогревает и поверхностные, и глубокие ткани организма. Кроме того, инфракрасная радиация, действуя на весь организм, интенсивно прогревает и ткань головного мозга, в котором располагаются нейроны центра терморегуляции. В связи с этим солнечный удар развивается быстротечно и чреват смертельным исходом.
Патогенез. Патогенез солнечного удара — комбинация механизмов гипертермии и собственно солнечного удара. Ведущими являются различные поражения ЦНС.
ГИПЕРТЕРМИЧЕСКИЕ РЕАКЦИИ.
Причиной гипертермических реакции являются непирогенные агенты. В основе развития гипертермических реакций обычно лежит временное преобладание теплопродукции над теплоотдачей. Механизмы терморегуляции организма при этом сохраняются.
ЛИХОРАДКА
Лихорадочная реакция — динамичный и стадийный процесс. По критерию изменения температуры тела выделяют три стадии лихорадки: I. подъёма температуры, II. стояния температуры на повышенном уровне и III. снижения температуры до значений нормального диапазона.
I. Стадия подъёма температуры тела характеризуется накоплением в организме дополнительного количества тепла за счёт преобладания теплопродукции над теплоотдачей.
II. Стадия стояния температуры тела на повышенном уровне характеризуется относительной сбалансированностью теплопродукции и теплоотдачи. Однако баланс этих двух процессов достигается уже на уровне, существенно превышающем долихорадочный. Именно это и поддерживает температуру тела на повышенном (по сравнению с долихорадочным периодом) уровне: интенсивная теплопродукция уравновешивается эквивалентной ей теплоотдачей.
III. Стадия снижения температуры тела до нормальной характеризуется постепенным снижением продукции лейкоцитарных пирогенных цитокинов.
ЛИХОРАДКА
Лихорадка – типовой патологический процесс, заключающийся во временном повышении температуры организма на действие пирогенных раздражителей в связи с перестройкой регуляции теплообмена на новый более высокий уровень. Лихорадка в своей основе является приспособительной реакцией, повышающей естественную резистентность организма.
Этиология. Непосредственной причиной лихорадки являются пирогенные (жаронесущие) вещества, или пирогены. По происхождению пирогены делят на экзогенные и эндогенные. Среди экзогенных выделяют инфекционные и неинфекционные пирогены, которые вызывают, соответственно, инфекционные и неинфекционные лихорадки [липополисахариды (ЛПС), липотейхоевые кислоты, пептидогликаны, термолабильные белковые вещества, нуклеиновые кислоты].
Неинфекционные (асептические) лихорадки. Первичные пирогены могут образовываться в организме не только в результате повреждения инфекционным процессом собственных тканей. Развитие асептической лихорадки вызывают первичные экзогенные и эндогенные неинфекционные пирогены после активации в организме макрофагальных и иных мезенхимальных клеток и продукции ими вторичных белковых эндопирогенов. Эндопирогены высвобождаются малигнизированными клетками миелоидного ряда при миелолейкозе, трансформированными в опухолевые клетки лимфомами, меланомами и другими злокачественными образованиями. Они появляются в случаях асептического травматического повреждения тканей, (травма, ушиб, раздавливание и т.п.), асептических воспалениях, гемолизе и т.д., развития ишемического или геморрагического некроза (инфаркты, кровоизлияния и т.п.), иммуноаллергических процессов и т.п., введения сывороток с лечебно-диагностическими целями, переливания крови и других белково-содержащих жидкостей и кровезаменителей. В этих случаях пирогены образуются клеточно-тканевыми структурами самого организма (компоненты комплемента – C3a, C5a, лейкотриены, антигены). При аллергических реакциях комплексы антиген-антитело, связываясь со специфическими рецепторами цитоплазматической мембраны, активируют гены, ответственные за синтез эндопирогенов
Кроме инфекционной и неинфекционной лихорадки, встречается еще одна группа гипертермий, которые могут быть вызваны без обязательного участия пирогенов. Они получили наименование лихорадоподобные состояния. К ним относят нейрогенные, эндокринные и лекарственные лихорадоподобные состояния. Нейрогенные «лихорадки» могут иметь центрогенное (повреждение структур ЦНС), психогенные лихорадоподобные состояния – функциональные нарушениях высшей нервной деятельности, рефлексогенные (при почечнокаменной, желчекаменной болезни и т.д.) происхождение. Последние всегда сопровождаются болевым синдромом. Эндокринные лихорадоподобные состояния возникают при некоторых эндокринопатиях (гипертиреозы, поражения гипоталамуса), а лекарственные – при использовании некоторых фармакологических препаратов (кофеин, эфедрин, гиперосмолярные растворы и т.д.).
Продолжительность лихорадки варьирует от нескольких часов до многих месяцев. По уровню повышения температуры во второй стадии выделяют следующие типы лихорадки: слабую (субфебрильную), когда температура повышается до 38о; умеренную (фебрильную – повышение до 38-39о); высокую (пиретическую – повышение до 39-41о) и чрезмерную (гиперпиретическую – повышение до 41о или чуть выше).
Температура тела человека подвержена суточным колебаниям: минимум утром в 4-6, максимум вечером в 17-19 часов. Этот температурный ритм в большинстве случаев сохраняется и при лихорадке. Следовательно, ход температурной кривой лихорадящего организма является результирующей суточных колебаний и стадийности развития самой лихорадочной реакции. При длительной гипертермии в зависимости от хода температурной кривой различают следующие основные типы лихорадки: постоянная (febris continua) – суточные колебания не превышают 1о С, ремиттирующая, или послабляющая (febris remittens) – суточные колебания более 1о С; перемежающая (febris intermittens) – суточные колебания от нормы до максимума; гектическая, или истощающая (febris hectica) – очень большие подъемы с быстрым спадом температуры, иногда повторяющиеся по несколько раз в течение суток; извращенная (febris inversa) – извращение суточного ритма с более высокими подъемами температуры по утрам; неправильная (febris atypica) – колебания температуры в течение суток без определенной закономерности; возвратная (febris recurrens) – возвращение лихорадки после нормализации температуры.
Биологическое значение лихорадки. Наиболее распространенным считается взгляд на природу лихорадки как на защитно-приспособительную реакцию организма. Лихорадка стимулирует выработку антител, интерферона, процессы фагоцитоза и является факторов повышения иммунологической реактивности организма. Она оказывает тормозящее влияние на развитие некоторых аллергических заболеваний.
Обладая защитными потенциями, лихорадка в значительно большей степени может при определенных условиях приобретать патогенное значение для организма. Именно вредоносное влияние лихорадки на организм должно интересовать медиков в первую очередь. Действительно, длительно лихорадящие состояния (при туберкулезе, бруцеллезе, раневом воспалении и т.п.) и гиперпиретические лихорадки приводят больных к крайней степени истощения и ослаблению многих физиологических функций.
Лихорадки.
Каждая лихорадка в своем развитии проходит три стадии: подъема (stadium incrementi), относительного стояния на повышенном уровне (stadium fastigii) и спада температуры (stadium decrementi). Разогревание организма в первой стадии может происходить в течение 0,5-3 час (малярия, крупозная пневмония и пр.) или нескольких суток (брюшной тиф и др.) по одному из нескольких вариантов теплообмена. При быстром подъеме температуры наблюдается усиленная теплопродукция в результате непроизвольного сокращения поперечно-полосатой мускулатуры (озноб) при одновременном снижении теплоотдачи путем спазма поверхностно расположенных сосудов, ограничения кровообращения на периферии тела, прекращения потоотделения, снижения интенсивности испарения пота. При медленном подъеме температуры возможно одновременное увеличение и теплопродукции, и теплоотдачи, но при условии превышения степени увеличения продукции тепла над его потерей.
В основе повышения температуры при лихорадке лежат физико-химические процессы, обусловливающие накопление в организме дополнительного количества тепла. В течение длительного времени существовало предположение, что саморазогревание организма при лихорадке является результатом «пожара обмена», то есть резкого увеличения теплопродукции при ограничении способности выделять избыток тепла. Однако в дальнейшем установили, что у лихорадящего больного теплопродукция может повышаться всего на 30-50% от исходного уровня; в то время как у здорового человека в условиях физического напряжения теплопродукция может увеличиваться на 40% и выше, не вызывая повышения температуры тела благодаря адекватному усилению теплоотдачи. Кроме того, лихорадящий организм сохраняет способность активно поддерживать установившуюся на повышенном уровне температуру, сопротивляясь внешнему перегреванию и переохлаждению. Именно поэтому относительно скоростное повышение температуры внутренней среды организма объясняется не столько химической терморегуляцией (наработкой тепла), сколько физической терморегуляцией – ограничением потери тепла. В механизмах ограничения потери тепла по ходу развития лихорадки существенное значение приобретает спазм сосудов кожи. Их сужение может ограничить потерю тепла до 70 % от нормальных величин.
Дополнительный прирост тепла возникает в результате активации механизмов сократительного и несократительного термогенеза. Эти механизмы подробно обсуждались выше. Здесь же подчеркнем, что терморегуляторный мышечный тонус, как и дрожь, являются специализированными формами мышечной деятельности, направленной на увеличение теплопродукции. Вначале формируется асинхронное сокращение мышечных волокон, что воспринимается как тоническое напряжение мышц – т.е. недрожательный термогенез. Дальнейшая активация мышц формирует сократительную деятельность отдельных двигательных единиц – возникает мышечная дрожь, т.е. дрожательный термогенез – непроизвольные залповые сокращения различных групп мышц, т.ч. и жевательных. Сокращение жевательных мышц сопровождается постукиванием зубов друг о друга. Дрожь способствует усилению мышечного метаболизма в 4-5 раз по сравнению с исходным уровнем. Поскольку при этом совершается незначительная механическая работа мышц, то почти вся производимая энергия выделяется в виде свободного тепла, которое приводит к саморазогреванию организма. У взрослых дрожь является основным и наиболее эффективным способом увеличения выработки тепла, чем сильнее озноб, тем интенсивнее поднимается температура тела. У грудных детей прирост теплопродукции обеспечивается за счет усиления недрожательного термогенеза, и лишь в исключительных случаях – за счет мышечной дрожи.
Вслед за срочным сократительным термогенезом включается отсроченный несократительный термогенез, обусловленный активацией механизмов химической терморегуляции – повышение тонуса симпатикуса, выброс адреналина, соматолиберина и СТГ, тиролиберина, ТТГ, Т3 и Т4, АКТГ и глюкокортикоидов, инсулина, глюкагона, паратгормона. Активация катаболических процессов обеспечивается известными цитокинами – ИЛ-1, ФНО и других. Сопряженные механизмы сократительного и несократительного термогенеза приводят к накоплению тепла в организме и повышению температуры внутренней среды до тех пор, пока она не достигнет уровня новой установочной точки температурного гомеостаза. На этом завершается первая стадия лихорадки.
Во второй стадии развития лихорадочной реакции теплоотдача в целом уравнивается с теплопродукцией. Этот баланс терморегуляторных процессов устанавливается на более высоком уровне, чем в норме, что и обеспечивает удержание повышенной температуры тела (ощущение жара).
В третьей стадии лихорадки «установочная точка» центра терморегуляции возвращается к исходному уровню, и восстанавливается нормальный температурный гомеостаз. Процесс теплоотдачи в этой стадии превышает теплопродукцию, уровень которой может не изменяться по сравнению с предыдущей стадией, или снижаться (усиливается потоотделение, расширяются периферические сосуды и т.п.). Выделяют два крайних типа развития третьей стадии лихорадки: постепенное, или литическое, и быстрое, или критическое падение температуры. При критическом падении, когда температура в течение 1-2 час может снизиться на 3-4о С, обычно наблюдается обильное потоотделение, сопровождаемое резким расширением периферических сосудов, что может привести к падению артериального давления и острой сосудистой недостаточности. Литическое снижение температуры протекает медленнее, в течение 12-16 час.
Пиротерапия
Искусственная гипертермия (пиротерапия) применяется в сочетании с другими воздействиями медикаментозного и немедикаментозного характера. Существует общая и местная пиротерапия.
Общая пиротерапия. Она проводится путем воспроизведения лихорадки с помощью очищенных экзопирогенов (например, пирогенала), стимулирующих синтез эндопирогенов. Субфебрильная или фебрильная лихорадка стимулирует адаптивные процессы в организме: специфические и неспецифические механизмы иммунологической реактивности (при некоторых инфекционных процессах – сифилисе, гонорее, постинфекционном артрите), пластические и репаративные процессы в костях, тканях и паренхиматозных органах (при их деструкции, повреждении, дистрофиях, после хирургических вмешательств).
Местная гипертермия. Она воспроизводится совместно с другими методами лечения для стимуляции регионарных механизмов защиты (иммунных и неиммунных), репарации и кровообращения. Регионарную гипертермию применяют при хронических воспалительных процессах, эрозиях и язвах кожи, подкожной клетчатки, а также при отдельных разновидностях злокачественных новообразованиях. Так, в онкологии гипертермию применяют в связи с ее несколькими противоопухолевыми эффектами:
· торможение митозов (особенно в S-фазе) в опухолевой клетке. Экспериментально установлено, что повышение температуры клеток карциномы с 43 ° до 44° C уменьшает их выживаемость в 1,5-2 раза;
· денатурация белков, липопротеидов и многих ферментов бластомных клеток, что сочетается с их гипергидратацией и разрушением;
· увеличение в опухолевой ткани глутатиона, повреждающего ДНК опухолевых клеток;
· повышение вязкости крови и нарушение микрогемоциркуляции в сосудах опухоли, нарастание в ней гипоксии, ацидоза, гиперосмии, снижающих жизнеспособность опухолевых клеток;
· потенцирование эффектов химио-, радио- и иммунотерапии.
ЭТИОЛОГИЯ
Причинами воспаления являются многочисленные флогогенные факторы, которые при своем действии на организм превосходят адаптивные возможности тканей. По происхождению все флогогенные раздражители объединяют в две большие группы – экзогенные и эндогенные. К экзогенным относят инфекционные и неинфекционные агенты. К инфекционным биологическим факторам мы будем относить действие бактерий, вирусов, грибов, паразитов и их токсинов; к неинфекционным биологическим агентам – животных и насекомых, действие чужеродных белков и т.п. Среди других экзогенных факторов выделяют (1) физические (электрический ток, лучистая энергия, ультразвук, тепло-холод и др.); (2) механические (давление, разрыв, инородное тело и др.); (3) химические (кислоты, щелочи и др.); (4) психогенные факторы (известны случаи возникновения и развития отдельных признаков воспаления под гипнозом).
К эндогенным флогогенным агентам относят камни и отложение солей (при уремии, желчно-каменной болезни, камнях почек, подагре и т.п.), продукты тканевого распада, злокачественные опухоли, тромбы и эмболы, иммунные комплексы антиген-антитело при их фиксации в органах, некоторые физиологические раздражители, а также сапрофитную микрофлору.
Общая картина событий при остром воспалении может быть представлена в виде последовательности:
1. АЛЬТЕРАЦИЯ:
а) Первичное повреждение;
б) Вторичное самоповреждение;
2. ЭКССУДАЦИЯ:
а) Сосудистые реакции:
1.Ишемия;
2. Артериальная гиперемия;
3. Смешанная гиперемия;
4. Венозная гиперемия;
5. Смешанный стаз.
6) Экстравазация жидкости:
в) Маргинация лейкоцитов;
г) Эмиграция лейкоцитов;
д) Внесосудистые процессы;
1. Хемотаксис;
2. Фагоцитоз;
3. ПРОЛИФЕРАЦИЯ:
1. Действие противовоспалительных механизмов;
2. Активация фибробластов;
3. Фиброплазия и ангиогенез репарация.
Патогенез внешних признаков острого воспаления . Полагают, что еще Цельс в 178 г. знал 4 основных признака воспаления: покраснение (rubor), боль (dolor), жар (calor), припухлость (tumor). Гален добавил пятый признак воспаления – нарушение функции (functio laesa).
1. Патогенез боли:
1) Ацидоз; 2) Образование брадикинина; 3) Отек; 4) Повышенное осмотическое давление; 5) Дизиония; 6) Механическое раздражение рецепторов в очаге воспаления.
2. Патогенез красноты:
1) Артериальная гиперемия; 2) Увеличенное количество функционирующих капилляров; 3) Повышение содержания окисленного гемоглобина в оттекающей крови.
3. Патогенез припухлости:
1) Артериальная и венозная гиперемия; 2) Эмиграция лейкоцитов; 2) Экссудация; 4) Отек; 5) Набухание тканевых элементов.
4. Патогенез жара:
1) Усиление обмена веществ в очаге воспаления; 2) Артериальная гиперемия; 3) Разобщение процессов дыхания и фосфорилирования; 4) Метаболический взрыв у лейкоцитов.
5. Патогенез нарушения функции;
1) Повреждение клеток; 2) Нарушение обмена веществ; 3) Нарушение кровообращения; 4) Боль; 5) Пролиферативные процессы.
Биогенные амины
К данной группе относятся гистамин, серотонин, а также полиамины (спермин, спермидин, путресцин, кадаверин). Гистамин поступает в очаг воспаления при дегрануляции мастоцитов (с их рекордным содержанием этого медиатора – до 3,5 пг на клетку), а также из базофилов (содержащих до 1 пг на клетку), тромбоцитов, эозинофилов и, в гораздо меньшей степени, гладкомышечных клеток и эндотелия. Он образуется при посредстве гистидиндекарбоксилазы из гистидина во всех клетках, но только тучные клетки накапливают его в значительных количествах в гранулах. Отметим, что дегрануляция возможна в ответ на различные стимулы:
> Связывание антигенов через гомоцитотропные иммуноглобулины и реагиновые рецепторы (при анафилактическом воспалении).
> Связывание фрагментов комплемента – анафилотоксинов С5a и С3а, в меньшей мере С4a (при обычном и анафилактическом воспалении).
> Нейропептиды диффузной эндокринной системы, например, вещество Р (при астме, вызванной физическими усилиями).
> Цитокины (ИЛ-1 и ИЛ-8) при гиперчувствительности замедленного типа (ГЗТ).
> Физические повреждения клеток (при механической или температурной травме).
> Агонисты простагландиновых рецепторов (изоцианаты синтетических красок и герметиков при аллергоидной «астме новостроек»).
> Никотиновую кислоту (при аллергоидной крапивнице в ответ на витамин РР).
При воспалении гистамин вызывает расширение артериол и повышение проницаемости венул. Он усиливает секрецию слизи, вызывает зуд и боль, способствует освобождению кининов и липидных медиаторов. Другие его эффекты перечислены в таблице 1. Следует отметить, что гистамин сужает крупные сосуды (что делает его участником анафилактического коронароспазма) и, подавляя функцию номотопного водителя сердечного ритма (через H1-рецепторы), способен вызвать аритмии, вплоть до фибрилляции (с участием Н2-рецепторов). Эти эффекты смертельно опасны при аллергическом и аллергоидном шоке.
Действие гистамина не продолжительно из-за его инактивации.
Серотонин у человека в тучных клетках отсутствует. В связи с этим считается, что его роль в воспалении у человека менее важна. Его источником могут быть тромбоциты, эозинофилы, а в кишечнике – энтерохромаффинные клетки. Медиатор образуется из триптофана и представляет собой 5-гидрокситриптамин.
В очагах воспаления освобождению серотонина способствуют агреганты и активаторы тромбоцитов, в частности, фактор активации тромбоцитов и тромбин, а также иммунные комплексы.
Серотонин имеет 4 типа рецепторов, действуя через которые он повышает проницаемость венул, способствует агрегации тромбоцитов, активирует моноциты. В то же время, он вызывает спазм гладких мышц в бронхах и неоднозначно влияет на сосуды. Серотонин способен оказать прямой вазоконстрикторный эффект, особенно на венулы, внося вклад в формирование стаза. На мозговые сосуды серотонин действует как вазоконстриктор (возможно, его эффект опосредован нервами) и участвует в патогенезе мигрени. Серотонин как нейромедиатор используется ядрами шва и участвует в регуляции сна и бодрствования, передаче сенсорной информации, формировании эмоций, а при системном гормональном действии стимулирует стероидогенез в надпочечниках.
Полиамины рассматриваются как противовоспалительные медиаторы и стимуляторы репарации, клеточные медиаторы ростового эффекта соматомединов.
Катехоламины тромбоцитарного происхождения участвуют в развитии спазма сосудов и восстановлении нарушенной сосудистой проницаемости. Подробнее роль биогенных аминов представлена в Таблице 1.
Полипептидные медиаторы
Большинство полипептидных медиаторов воспаления присутствует в биологических жидкостях организма до начала воспаления в неактивной форме и вступает в действие в результате каскадного протеолиза.
Условно их можно подразделить на несколько групп: контактную систему плазмы крови, лейкокинины, цитокины, ферменты и антиферменты, катионные не ферментативные белки, транспортные и распознающие белки, нейропептиды, факторы роста.
Транспортные белки-участники воспаления – это церулоплазмин, транскобаламин, трансферрин, ферритин, которые, в основном, имеют значение как компоненты антиоксидантных и прооксидантных механизмов тканей.
Компонентами сторожевой системы являются плазменные протеазы: комплемент, свертывающая система, система фибринолиза и кининовая система. Они функционально едины, тесно связаны макрофагальным происхождением своих белков, имеют общее свойство «плавающих регуляторов» (в крови имеются их проактиваторы), работают по каскадному принципу, взаимно запускают друг друга и имеют общие эффекторы. Ядром сторожевой полисистемы служат 4 белка:
1. Фактор Хагемана (XII фактор свертывания крови);
2. Высокомолекулярный кининоген;
3. Плазменный прекалликреин;
4. XI фактор свертывания крови.
Система комплемента
Ранее предполагалось, что существует единственный термолабильный компонент плазмы, опосредующий литическое действие антител на бактерии. К настоящему времени идентифицировано 13 белков системы комплемента и 7 ингибиторов. Эти регуляторы циркулируют в неактивной форме (за исключением фактора D, который в активном виде присутствует в плазме в малых количествах), самособираются в ответ на определенные сигналы, активируют друг друга (причем служат при этом сериновыми протеазами и/или взаимными рецепторами), а в результате осуществляют несколько важных эффектов:
> Лизис мишеней, активирующих комплемент;
> Опсонизация объектов, фиксирующих факторы комплемента;
> Хемотаксис и усиление фагоцитоза;
> Активация лейкоцитов и опосредование их адгезии;
> Регуляция иммунного ответа;
> Освобождение медиаторов воспаления.
Белки комплемента условно подразделяются на факторы классического пути активации (обозначаются буквой С с ответствующими индексами – С1, С2, С4), факторы альтернативного пути активации (В, D), терминальные компоненты комплекса мембранной атаки (С5, С6, С7, С8, С9), а также усилители и ингибиторы комплемента (Р, Н, I, С4dp, DAF, МСР, НRF, С1INН и др.). Особняком стоит центральный фактор всей системы С, входящий в оба пути активации комплемента и участвующий в реализации практически всех его функций.
Медицина нередко встречается с наследственными и приобретенными дефектами системы комплемента. Эти состояния разнообразны и могут быть вызваны как наследственными мутациями (дефициты С1INH, Р, I), так и приобретенными состояниями, но их клинические проявления, как правило, сходны и включают снижение устойчивости к бактериальным инфекциям из-за нарушения литических и опсонизирующих функций комплемента, и развитие иммунокомплексных заболеваний (ИК-синдромов) из-за помех в клиренсе иммунных комплексов.
Тотальная активация комплемента происходит при контакте плазмы с мембранами ионо-обменников искусственной почки и других устройств для экстракорпоральной терапии. Аналогичные осложнения могут быть и у пациентов с эндопротезами сосудов. Результатом является системное действие анафилотоксинов и медиаторов активированных комплементом лейкоцитов, что формирует постперфузионный синдром, сопровождаемый лихорадкой, шоком, внутрисосудистым гемолизом, лейкопенией и гипокомплементемией потребления, кровоточивостью по капиллярному типу. Синдром исключается только в том случае, если все поверхности, с которыми контактирует кровь (плазма), будут неактивирующими.
Системная активация комплемента происходит при бактериемии грамотрицательными возбудителями, особенно, сальмонеллами, менингококками, пневмококками, гемофильной палочкой, при вирусемии возбудителями геморрагических лихорадок. Это важный элемент патогенеза инфекционно-токсического шока (шокового легкого).
При ожоговой болезни в системном кровотоке появляется избыток активных фрагментов комплемента, обусловливающих, наряду с прочими факторами, развитие ожогового шока и респираторного дистресс-синдрома в легких.
При остром панкреатите и травмах поджелудочной железы панкреатические протеазы активируют сторожевую полисистему крови, проникая в системный кровоток. Это ведет не только к системному действию кининов, но и к продукции анафилотоксинов. У больных может развиться тяжелый коллапс, диссеминированное внутрисосудистое свертывание крови и плюриорганная недостаточность, в том числе, шоковое лёгкое.
Очень велика роль расстройств функций комплемента в развитии нефропатий. Все нефриты, в том числе, инфекционные стрептококковые протекают с гипокомплементемией. При мембранозно-пролиферативной форме хронического диффузного гломерулонефрита в крови появляются аутоантитела к активной форме конвертазы альтернативного пути комплемента. Аутоантитела к конвертазе классического пути комплемента присутствуют при остром постстрептококковом нефрите и системной красной волчанке. Эти аутоантитела (нефритогенные факторы) блокируют освобождение ингибитором Н фактора С3 из состава конвертазы, и происходит снижение плазменной концентрации этого фактора. В результате нарушается клиренс иммунных комплексов, и они откладываются в клубочках почек, активируется комплементзависимый лизис эндотелия и других тканей и ослабевает устойчивость к гноеродной, в том числе, менингококковой инфекции. Нефритогенный фактор характерен и для парциальной липодистрофии, зачастую сопровождаемой дефицитом С3 и гломерулонефритом. При любых видах нефротического синдрома факторы комплемента, особенно, В, Р и С4, теряются с мочой, что обусловливает вторичную гипокомплементемию и иммунодефицит по отношению к бактериальной инфекции. При цитотоксической форме аутоиммунного гломерулонефрита (подострый злокачественный гломерулонефрит с «полулуниями», гломерулонефрит при синдроме Гудпасчера) комплемент опосредует лизис ткани клубочков под воздействием аутоантител к компонентам их базальной мембраны.
При СПИДе имеется дефицит ряда факторов комплемента на фоне значительного избытка в крови С3а. В связи с иммуносупрессивным действием этого анафилотоксина предполагается, что его накопление вносит вклад в развитие иммунологической недостаточности у таких больных.
Содержание многих факторов комплемента снижено по сравнению с взрослыми у новорожденных и, особенно, недоношенных детей, при голодании и печеночной недостаточности. Поэтому во всех этих случаях понижена антибактериальная резистентность.
Липидные медиаторы
Источниками липидных медиаторов является арахидоновая кислота. Свободная арахидоновая кислота окисляется в двух альтернативных путях образования липидных медиаторов. Один из этих путей контролируется ферментом циклооксигеназой и приводит к образованию простаноидов, к которым принадлежат простагландины и тромбоксаны. Циклооксигеназа блокируется салицилатами, например, аспирином, а также индометацином, ибупрофеном и другими нестероидными противовоспалительными агентами.
Другой путь осуществляется при участии фермента липооксигеназы и активирующего этот энзим мембранного белка FLAP. Он ведет к эйкозаполиеновым кислотам и лейкотриенам. Получены избирательные ингибиторы липооксигеназного пути арахидонового каскада, например зилевтон.
Циклооксигеназный путь начинается с появления циклической эндоперекиси арахидоната – просгагландина D2, (2 обозначает количество двойных связей в боковой цепи).
Затем формируется простагландин Н2 (PgH2,), а из него – наиболее биологически активные PgD2, PgE2, PgF2, а в эндотелии и гладкомышечных клетках сосудистой и бронхиальной стенки – также и циклический PgI2 ,известный как простациклин.
Включение в кольцо эндоперекисей с помощью фермента тромбоксансинтетазы дополнительного кислородного атома позволяет преобразовать PgH2, в тромбоксан А2 (ТхА2), что, главным образом, происходит в мегакариоцитах и тромбоцитах, а в эндотелии осуществляется менее активно. Другим медиатором этого ряда служит ТхВ2, образуемый из ТхА2. Добавим, что, помимо простагландинов, гидроксиэйкозанолиеновых кислот, тромбоксанов, простациклина, лейкотриенов, при воспалении ряд клеток, в том числе, нейтрофилы вырабатывают еще одну группу липидных медиаторов – тригидроксипроизводные арахидоновой кислоты липоксины. При остром воспалении наиболее велика роль простагландина Е, лейкотриена В4, 5-гидроксиэйкозатетраеновой кислоты и фактора активации тромбоцитов. Особенно большое значение придают последнему фактору. Ацетилглицериновый эфир фосфорилхолина образуется из материала плазматических мембран под влиянием фосфолипазы А. Несмотря на простоту своей структуры – это мощнейший и разносторонний посредник воспалительных реакций. Достаточно упомянуть, что блокаторы его образования обрывают практически все симптомы острого воспаления во многих экспериментальных моделях. Самым сильным стимулятором выработки фактора активации тромбоцитов является тромбин. Однако цитокины, в частности, ИЛ-1, кинины, лейкотриены, внеклеточная АТФ и гистамин тоже способны повышать его продукцию.
Полисахаридные медиаторы
Полисахаридные медиаторы воспаления – это гликозаминогликаны (гепарин, хондроитин-сульфаты, гепаран-сульфат, дерматан-сульфат). Они вырабатываются многими клетками: фибробластами, гладкомышечными элементами, эндотелием, макрофагами.
Спектр гликозаминогликанов, синтезируемых различными клетками, отличается. Для гепарина основными продуцентами служат базофилы и мастоциты, для гепаран-сульфата – эндотелий и т. д.
Большинство эффектов полисахаридных медиаторов связано, так или иначе, с противовоспалительным действием.
Гепарин и, особенно, гепаран-сульфат являются сильными антикоагулянтами и антиагрегантами. Они, а также дерматан-сульфат препятствуют тромбообразованию и фибринообразованию, способствуют фибринолизу, стимулируя выделение эндотелием активатора плазминогена. Гепаран-сульфат действует через тканевой рецептор антитромбин III, гепарин – через кофактор II. Взаимодействуя с коллагеном и адгезивными белками, гликозаминогликаны принимают участие в самосборке ткани при фиброплазии и регенерации. Как фактор миграции эндотелиоцитов гепарин способствует ангиогенезу.
ЗАЩИТНО-ПРИСПОСОБИТЕЛЬНОЕ ЗНАЧЕНИЕ ВОСПАЛЕНИЯ 1. Воспалительная реакция выработалась в процессе эволюции. Защитный характер воспаления состоит в том, что организм, включая воспаление как защитную реакцию, активно локализует очаг повреждения с помощью так называемого «защитного вала» (прекращение оттока крови и лимфы, лейкоцитарная инфильтрация ткани и т.д.), препятствуя тем самым распространению патогенного раздражителя по организму. Чем более местно протекает эта общая реакция, тем благоприятнее для организма ее исходы.
2. Наряду с отграничительной функцией воспаление создает условия для инактивации (уничтожения) тем или иным способом (фагоцитоз, ферментолиз, иммунный цитолиз, образование экссудата, адсорбции токсинов и т.п.) патогенного раздражителя (и поврежденных им тканевых структур) непосредственно в регионе его действия.
3. Очаг воспаления обладает дренажной функцией, т.к. создает условия для выделения из зоны повреждения патогенных раздражителей и продуктов распада тканей во внешнюю среду.
4. В очаге воспаления формируются условия для мобилизации и защитных сил организма специфического и неспецифического характера.
30.Противовоспалительные и провоспалительные гормоны. Роль нервной и эндокринной системв развитии воспаления.Регуляция воспаления осуществляется с помощью гормональных, нервных и иммунных факторов.
Известно, что некоторые гормоны усиливают воспалительную реакцию - это, так называемые, провоспалительные гормоны (минералокортикоиды, соматотропный гормон гипофиза, гипофизарный тиреостимулин, альдостерон). Другие, наоборот, уменьшают ее. Это противовоспалительные гормоны, такие как глюкокортикоиды и адренокортикотропный гормон (АКТГ) гипофиза. Среди системных регуляторов известное значение для затухания воспаления, особенно в условиях противовоспалительной терапии, имеют глюкокортикоидные гормоны. Их противовоспалительное действие связано со стимуляцией продукции макрофагальных антифосфолипаз, тормозящих арахидоновый каскад, а также с подавлением экспрессии генов интерлейкинов и индукцией апоптоза лимфоцитов и эозинофилов. Глюкокортикоиды снижают интенсивность трансцитоза и блокируют в клетках-участниках воспаления некоторые локомоторные функции, зависящие от цитоскелета. Они тормозят активность и экзоцитоз коллагеназ и других протеаз. Большое значение для разрешения острой фазы воспаления имеет функция лимфатической системы.
Холинергические вещества, стимулируя выброс медиаторов воспаления, действют подобно провоспалительным гормонам, а адренергические, угнетая медиаторную активность, ведут себя подобно противовоспалительным гормонам.
Удаление щитовидной железы ослабляет развитие воспаления, а введение тироксина усиливает воспалительную реакцию.
Некоторое влияние на проницаемость кровеносных капилляров оказывают половые гормоны. Эстрогены заметно подавляют активность гиалуронидазы. Удаление поджелудочной железы усиливает тяжесть воспалительной реакции: фагоцитарная активность лейкоцитов в этих условиях снижается.
О. Синдром приобретенного иммунодефицита (СПИД): этиология, патогенез, проявления, профилактика.
Одним из наиболее клинически значимых ИДС этой группы является синдром приобретенного иммунодефицита (СПИД, AIDS ). В истории медицины, пожалуй, найдется немного болезней, вызвавших столько тревог, сомнений и толков, как пресловутый СПИД. Первое сообщение о новом заболевании иммунной системы человека появилось в США в 1979 году, но только спустя 2 года, в 1981 г., болезнь стали регистрировать как самостоятельную нозологическую единицу. Заболевание приняло характер эпидемии, и количество больных прогрессивно увеличивается во всех странах. В России число новых случаев заболеваний, выявленных только весной 1997 г., превысило показатель всего 1996 года, и с 1997 г. в нашей стране объявлена эпидемия по СПИДу.
Этиология. Возбудителями СПИДа являются вирусы, относящиеся к группе ретровирусов, или герпесвирусов. Ведущая роль принадлежит вирусу иммунодефицита человека (ВИЧ -HTV). Описаны три формы ВИЧ (1, 2, 3). Заболевания в Европе, Америке, Австралии и Центральной Африке вызываются вирусом ВИЧ-1, а в Западной Африке - вирусом ВИЧ-2. ВИЧ проникает в организм человека тремя четко установленными путями: (1) гемотрансфузионный (при переливании крови или ее продуктов, включая использование коллективных шприцов наркоманами или их недостаточную стерилизацию, и клеток при пересадке тканей), (2) половой (во время полового акта), (3) перинатальный (антенатальный – в период беременности, когда плод больной женщины может инфицироваться через плаценту, интранатальный – при продвижении плода по инфицированным родовым путям роженицы). Среди других путей заражения отметим инструментальный (повторное использование загрязненных инструментов), трансплантационный (пересадка зараженных органов), молочный (заражение ребенка инфицированным материнским молоком), профессонально-бытовое заражение (через поврежденные кожные покровы и слизистые медперсонала, контактирующего с биологическими жидкостями больных СПИДом).
Патогенез. Вирус внедряется в клетки, имеющие на мембране рецепторы типа CD4, к которым ВИЧ проявляет высокий аффинитет. Наиболее богаты такими рецепторами Т-лимфоциты хелперы (Th, или Т4, по номенклатуре ВОЗ), макрофаги, дендритические клетки лимфоидной ткани и кожи (клетки Лангерганса), клетки глии, нейроны головного мозга и клетки слизистой желудочно-кишечного тракта. Спустя 6-9 недель, реже 6-9 месяцев после инфицирования появляются антитела к антигенам, представленным в капсиде ВИЧ в виде протеинов с молекулярной массой 120, 41, и 24 D (p 120, p 41 и p 24 D), которые могут быть выявлены иммунологическими методами. ВИЧ внедряется в цитоплазматическую мембрану клетки-мишени и изменяет ее свойства таким образом, что дальнейшее проникновение его значительно облегчается.
Оболочка вируса разрушается, и фрагменты его РНК и фермент обратная транскриптаза полностью высвобождаются. С помощью обратной транскриптазы на этой РНК, как на матрице, синтезируются белковые компоненты и копии ДНК вируса, которые далее проникают в ядро клетки-мишени и встраиваются в ее геном. В виде провируса ВИЧ находится в клетке в течение всей ее жизни.
Измененная генетическая информация в ядре клетки-мишени длительное время может не приводить к нарушениям в иммунном статусе. Эта дремлющая ВИЧ-инфекция активируется в процессе активации самой клетки хозяина – носителя вируса, и тогда появляются развернутые симптомы болезни в различных клинических формах.
Роль пусковых механизмов в реактивации провируса выполняют как специфические, так и неспецифические (поликлогальные) активаторы лимфоидных клеток: вирусы герпеса, Эпштейна-Барра, цитомегаловирус и другие. В качестве кофакторов могут выступать патогенные и непатогенные агенты, включая медикаментозные вмешательства, например, необоснованное лечение иммуностимуляторами, ультрафиолетовое, рентгеновское и другие формы лучевого воздействия, физические и психо-эмоциональные перегрузки, алкоголь и даже беременность.
По завершении процесса самосборки вирусные частицы отторгаются от клетки, попадают в межклеточную жидкость, лимфу, кровь, атакуя и приводя к гибели все новые и новые клетки, имеющие СD-4 (Т4) рецепторы. В наибольшей степени страдают Т-хелперы (Th или Т4-лимфоциты), что ведет к значительному уменьшению их числа, развитию лимфоцитопении и дефициту интерлейкина-2 (ИЛ-2). Когда число лимфоцитов падает ниже критического (менее 600 клеток/мл), формируется ИДС по клеточному типу, которое характеризуется развитием оппортунистической инфекции, вызываемой Pneumocystis carinii, Candida albicans, Mycobacterium avium-intracellular, Herpes zoster и т.п. Из других проявлений ВИЧ-инфекции отметим лихорадку, припухлость лимфатических узлов (вплоть до лимфоаденопатии), сыпь, менингеальные симптомы, ночные потоотделения, потеря в весе, тромбоцитопения, деменция, параличи.
На 80-90% снижается количество и функциональная активность естественных клеток-киллеров (NK). Число моноцитов и макрофагов может не изменяться, но целый ряд их функциональных свойств значительно расстраивается, в частности, страдает презентация ими антигена Т- и В-лимфоцитам. Во время ранней фазы ВИЧ-инфекции наблюдается выраженная поликлональная экспансия В-лимфоцитов, результатом чего является повышенное содержание в сыворотке крови иммуноглобулинов G-, M- и A-классов. На заключительных этапах AIDS содержание этих иммуноглобулинов резко падает, особенно в отношении титра антител к p 120 и p 24.
Это создает предпосылки для развития сопутствующих привходящих заболеваний - инфекций, лимфоретикулярных опухолей (В-клеточная лимфома, саркома Капоши), опухолей из эндотелиальных клеток, которые характеризуются выступающими пурпурными пятнами на коже, неспособности организма к формированию аллергических реакций замедленного типа. Таким образом, человек, инфицированный ВИЧ, становится беззащитным перед микробами, которые не представляют угрозу для здорового организма.
Клинические варианты заболевания СПИД.
1. Преимущественное поражение легких. Легочный тип поражения характеризуется развитием пневмоний, вызванных сопутствующей инфекцией, чаще пневмоцистами.
2. Преимущественное повреждение ЦНС - развитие энцефалита и менингита.
3. Преимущественное поражение желудочно-кишечного тракта. Эта форма характеризуется признаками поражения желудочно-кишечного тракта, в первую очередь диареей, вызываемой у 90-95% больных преимущественно оппортунистической инфекцией.
4. Лихорадочный тип. Характеризуется возникновением длительной лихорадки, не связанной с другими заболеваниями, сопровождающейся не мотивированным значительным снижением массы тела (более чем на 10%), слабостью, профузным потоотделением в ночные часы.
При всех формах течения СПИД отмечается повышенная склонность к образованию опухолей.
Методов эффективной терапии СПИД не существует. Лечебные мероприятия направлены на:
1. Блокаду размножения ВИЧ (подавление репликации его нуклеиновой кислоты путем ингибирования ревертазы дидеоксинуклеозидами, в частности азидотимидином; супрессия процессов трансляции и «сборки» вируса);
2. Подавление и профилактику инфекций и опухолевого роста;
3. Восстановление иммунной компетентности организма (введение препаратов тимуса, ткани костного мозга, интерлейкина-2).
Предложен метод очистки крови с помощью иммуноплазмосорбции и иммуноплазмафереза, причем при таких процедурах иммуносорбент и, соответственно, иммунополистирольная пленка снабжены ковалентно прикрепленным к ним CD4-белком - это проявляет более высокое сродство с ретровирусом ВИЧ.
И поскольку способов излечения больных СПИДом не существует, есть только два метода эффективной защиты населения от инфекции. Первый - связан с поголовной диспансеризацией населения на предмет выявления явных и скрытых носителей СПИДа с введением в стране карантинных служб. Второй - с изменением менталитета общества, постепенным повышением его половой культуры, появлением в его среде новых норм морали.
Вопрос 53.
Вопрос 54.
ГИПОГИДРАТАЦИИ
Гипогидратация возникает в следующих случаях:
1) вследствие нарушения поступления воды в организм (водное голодание, нарушение глотания, коматозное состояние и др.);
2) вследствие повышенной потери воды (кровопотеря, полиурия, понос или рвота, гипервентиляция, усиленное потоотделение, потеря биологических жидкостей с экссудатом или с обширных раневых поверхностей).
При обезвоживании теряется в первую очередь внеклеточная жидкость и ионы натрия, а при более тяжелой его степени – ионы калия и внутриклеточная жидкость. Крайняя степень обезвоживания называется эксикозом и считается наиболее тяжелой формой расстройства водного обмена.
В целом обезвоживание влечет за собой уменьшение объема циркулирующей крови – гиповолемию, сгущение крови и повышение ее вязкости, тяжелые нарушения кровообращения, микроциркуляции, коллапс. Нарушения кровообращения ведут к развитию гипоксии, в первую очередь, ЦНС. Гипоксия клеток ЦНС может сопровождаться помрачением сознания, комой, нарушением функций жизненно-важных центров. Одновременно гипогидратация сопровождается развитием компенсаторных реакций. Гиповолемия и снижение в результате этого почечного кровотока вызывают избыточную продукцию АДГ и альдостерона, под действием которых усиливается реабсорбция в дистальных отделах нефрона воды и ионов натрия. Диурез может уменьшиттся в 5 раз до уровня облигатного количества мочи, которое еще не вызывает нарушений выведения азотистых шлаков. Но дальнейшее концентрирование мочи, когда ее плотность увеличивается до 1040 и выше, приводит к развитию канальцевого ацидоза и гибели тубулярного аппарата.
1. ГИПЕРОСМОЛЯРНАЯ ГИПОГИДРАТАЦИЯ
Развивается вследствие потери организмом жидкости, обедненной солями, т.е. потеря воды превышает потерю электролитов; это обезвоживание возникает в связи с первичной абсолютной нехваткой воды – водного истощения, десикации, эксикоза. Причинами гиперосмолярной дегидратации могут быть следующие факторы:
1. Алиментарное ограничение поступления воды в организм:
а) затруднения глотания вследствие сужения пищевода, опухоли и др.;
б) у тяжелобольных в коматозном и критическом состояниях, тяжелые формы истощения;
в) у недоношенных и тяжелобольных детей;
г) отсутствие чувства жажды при некоторых формах заболеваний головного мозга (например, микроцефалия).
2. Избыточные потери воды через легкие, кожу, почки:
(а) гипервентиляционный синдром, (б) лихорадка, (в) усиленное потоотделение при повышении температуры окружающей среды, (г) искусственная вентиляция легких, которую проводят неувлажненной дыхательной смесью, (д) отделение больших количеств слабо концентрированной мочи (при несахарном диабете).
3. Потери гипотонической жидкости с обширных обожженных и травмированных поверхностей тела.
4. Гипергликемия.
Патогенез. Потеря воды, гемоконцентрация приводят к увеличению содержания натрия (до 160 ммоль/л) и повышению осмотического давления во внеклеточном прстранстве (выше 300 мОсм/л). Повышение осмотического давления в свою очередь влечет за собой перемещение части воды из клеток в околоклеточное пространство. Возникает гипогидратация клеток, эксикоз.
Гемоконцентрация сопровождается увеличением показателя гематокрита, возрастаним содержания белка в плазме (относительная гиперпротеинемия) и ведет к развитию типовых нарушений. Обезвоживание внеклеточного сектора приводит к развитию гиповолемии и артериальной гипотензии. В результате гиповолемии развивается циркуляторная гипоксия, которая усиливает расстройства микроциркуляции, внутриваскулярные расстройства микроциркуляции сопровождаются нарушениями реологических свойств крови – сгущением, повышением вязкости, развитием стаза и сладж-синдрома. Экстрасосудистые расстройства микроциркуляции ведут к нарушениям тока межклеточной жидкости и последующей гипоксии и завершаются дезорганизацией метаболических процессов в тканях, а именно: протеолизом (распад белка), гиперазотемией (увеличением сордержания остаточного азота более 40 мг% или 28,6 ммоль/л), аммиака (вследствие избытка его образования в тканях и ограничения утилизации печенью), мочевины (нарушение функции почек – ретенционная гиперазотемия), гипертермией, возникновением мучительного чувства жажды. В зависимости потери определенных ионов развивается либо ацидоз (потеря натрия, бикарбонатов), либо алкалоз (потеря калия, хлора).
2. ИЗООСМОЛЯРНАЯ ГИПОГИДРАТАЦИЯ.
Это такой вариант нарушения водного баланса, в основе которого лежит эквивалентное уменьшение объема жидкости и электролитов во внеклеточной среде. При этом возникает солевой дефицит, осложненный потерей соответствующего количества жидкости. Наиболее частыми ее причинами являются:
(1) острые кровотечения, (2) полиурия, (3) острая патология системы пищеварения:
а) стеноз привратника; б) острая бактериальная дизентерия; в) холера; г) язвенный колит; д) высокая тонкокишечная непроходимость; е) тонкокишечный свищ.
При изоосмолярной гипогидратации потеря воды из внеклеточной жидкости приводит, в первую очередь, к нарушениям гемодинамики, сгущению крови (ангидремия). При быстром обезвоживании организма потеря воды плазмы инициирует движение жидкости из клеток во внеклеточное пространство. Сильная кровопотеря (от 750 до 1000 мл за сутки) приводит к перемещению воды из внеклеточного пространства в сосуды, восстанавливая объем циркулирующей крови.
Нарушения функций. Потеря жидкости, близкой по составу к внеклеточной и плазме, приводит к тяжелым нарушениям функций (организм теряет натрий, хлор, воду) – прогрессивно снижается масса тела, падает артериальное и венозное давление, снижается минутный объем сердца, нарушается деятельность ЦНС (расстройство сознания, прострация, кома), нарушаются функции почек (олигурия, вплоть до анурии), могут возникать гипотензия и шок. Если дегидратация обусловлена потерей большого количества желудочного сока (например, вследствие рвоты), то возникает гипохлоремия и метаболический алкалоз (повышается содержание бикарбонатов плазмы).При диарее уменьшается количество бикарбонатов, а сопутствующие гипотензия и нарушения кровообращения приводят к развитию метаболического ацидоза вследствие нарушения периферического кровообращения и гипоксии.
II. 1. 3. ГИПООСМОЛЯРНАЯ ГИПОГИДРАТАЦИЯ.
Она возникает вследствие потери жидкости, обогащенной электролитами. Такое состояние обычно является результатом перехода острой дегидратации в хроническую (хронический дефицит электролитов). Наиболее частыми причинами гипоосмолярной гипогидратации являются:
1. Потери через желудочно-кишечный тракт: (а) долго незаживающие свищи желудка, кишечника, поджелудочной железы, (б) рвота, поносы и другие диспептические расстройства;
2. Потери через почки: (а) полиурия с высокой осмотической плотностью мочи, (б) осмотический диурез, (в) болезнь Аддисона (дефицит альдостерона), (г) идиопатический ацидоз новорожденных (у детей до шести месяцев нет карбоангидразы, вследствие чего и нарушается реабсорбция натрия);
3. Потери через кожу (обильное потоотделение у рабочих горячих цехов, тяжелая физическая работа);
4. Возмещение изотонических потерь жидкостей организма растворами, не содержащими электролитов (неправильная коррекция), а также прием большого количества пресной воды;
5. «Синдром больной клетки» – перемещение натрия из внеклеточного пространства в клетки.
Патогенез. Потеря почками жидкости и в еще большей мере электролитов желудочно-кишечным трактом ведет к гипоосмолярности внеклеточной жидкости (осмотическое давление внеклеточного сектора менее 300 мОсм/л) и в случае выраженной гипоосмолярной гипогидратации возможно вторичное перемещение воды, которая начинает поступать из внеклеточного сектора в клетку. Это может привести к дальнейшему увеличению степени внеклеточной гипогидратации при одновременном развитии внутриклеточного отека (внутриклеточная гипергидратация).
Следует учитывать потерю организмом определенных ионов, в частности натрия и калия. Потеря калия сопровождается жаждой и переходом воды из клеток в околоклеточные пространства. Компенсация теряемого натрия осуществляется за счет внеклеточной жидкости. Поэтому здесь на первый план выступают нарушения кровообращения (гиповолемия без развития жажды). Потеря натрия пищеварительными соками сопровождается ацидозом, а калия - алкалозом.
Проявления синдрома общей дегидратации.
| Жажда | Появляется даже при небольшом дефиците воды при гипернатриемии. Дефицит 3–4 л воды вызывает мучительную жажду. |
| Сухость кожи и слизистых оболочек | Особенно в подмышечной и паховой областях. |
| Гипосаливация | При длительном процессе способствует развитию воспаления в ротовой области и кариеса. |
| Язык | Гладкий, красный, с глубокими морщинами. |
| Глазные яблоки | Запавшие, мягкие при надавливании. |
| Тургор тканей (кожи, мышц) | Снижен. |
| Почечный кровоток | Снижен. Могут появиться признаки почечной недостаточности на фоне олиго– и анурии (азотемия, ацидоз и др.). |
| Процессы переваривания и всасывания питательных веществ | Угнетены, т.к. нарушается выделение соков. |
| Неврологические симптомы | Слабость, вялость, апатия, сонливость или возбуждение. |
| Температура тела | Повышена. |
| Масса тела | Снижена. |
ГИПЕРГИДРАТАЦИЯ
Гипергидрия возникает либо вследствие избыточного поступления в организм воды, либо неэффективного ее выведения, либо комбинации того и другого. Подобное наблюдается:
1. При дефиците тироксина и/или тиреотропного гормона аденогипофиза;
2. Избытке антидиуретического гормона;
3. Гиперальдостеронизме.
Возможны три варианта гипергидрий: изоосмолярня, гипоосмолярная и гиперосмолярная гипергидратации.
1. ИЗООСМОЛЯРНАЯ ГИПЕРГИДРАТАЦИЯ.
Она воспроизводится в эксперименте путем введении в организм избыточного объема физиологического раствора. Развивающаяся при этом гипергидрия носит временный характер. Осмотическое давление во внеклеточной жидкости не изменено и составляет 300 мОсм/л. Внеклеточное пространство может увеличиваться на несколько литров без признаков отека. Видимые отеки возникают при накоплении в организме около 3 литров жидкости.
Патогенез. Вследствие гипергидратации внутрисосудистого сектора происходит снижение показателя гематокрита и концентрации белков плазмы крови (относительная гипопротеинемия). Это сопровождается снижением онкотического давления, что облегчает транспорт воды из внутрисосудистого сектора в ткани, и увеличением диуреза вследствие нарастания фильтрационного давления и рефлекторного снижения секреции АДГ (Рис.2). Несоответствие скорости образования мочи степени гипергидратации ведет к образованию отеков.
При изоосмолярной гипергидратации образование отеков первично. Как правило, отеки связаны с увеличением реабсорбции натрия в почках вследствие вторичного альдостеронизма, который возникает в период формирования отеков. При этой форме гипергидратации организм переполнен водой, но не может ее использовать.
Наиболее частыми причинами изоосмолярной гипергидратации являются:
1. Сердечная недостаточность (миокардическая форма);
2. Патология почек;
3. Цирроз печени;
4. Прием и введение солевых изотонических растворов при сниженной выделительной функции почек (олигуия, анурия);
5. Опухоли коркового вещества надпочечников (альдостерома).
2. ГИПООСМОЛЯРНАЯ ГИПЕРГИДРАТАЦИЯ
Она возникает вследствие первичного избытка воды – «водная интоксикация». Причинами гипоосмолярной гипергидратации являются:
1. Задержка диуреза вследствие почечной недочстаточности;
2. Осложнение инфузионной терапии изотоническим (5%) раствором глюкозы;
3. Избыточный прием жидкости через рот или при многократной ирригации толстого кишеччника;
4. Избыточная продукция АДГ:
а) послеоперационные состояния;
б) болезнь Пархона;
в) боль, страх;
г) тяжелая мышечная работа;
5. Увеличение образования эндогенной воды при распаде тканей;
6. Бессолевая диета;
7. Применение лекарств, увеличивающих выведение натрия.
Патогенез. Гипоосмолярная гипергидратация формируется одновременно в клеточном и внеклеточном секторах и потому относится к тотальной гипергидрии. Внутриклеточная гипоосмолярная гипергидратация сопровождается грубыми нарушениями электролитного обмена и кислотно-щелочного баланса (снижение содержания ионов натрия в плазме), а также уменьшением величины мембранного потенциала клеток. При водном отравлении могут наблюдаться тошнота, рвота, судороги, кома («гипоосмолярная кома»), гиперрефлексия.
3. ГИПЕРОСМОЛЯРНАЯ ГИПЕРГИДРАТАЦИЯ.
Она может возникнуть в результате вынужденного приема морской или соленой воды в качестве питьевой, результатом чего является быстрое нарастание концентрации электролитов во внеклеточных пространствах - острая гиперосмия (осмолярность более 300 мОсм/л) вследствие того, что плазмолемма не пропускает избытка ионов в цитоплазму. Однако она не может удерживать в клетке воду, и последняя перемещается в межклеточные пространства. В результате нарастает внеклеточная гипергидратация, что несколько снижает степень гиперосмии. Одновременно из-за потери воды в клетках развивается обезвоживание (внутриклеточная дегидратация). Подобный тип нарушений сопровождается развитием таких же симптомов, как и при гиперосмолярной гипогидратации, ведущим из которых является нарастающая жажда, заставляющая человека вновь и вновь принимать соленую воду.
Увеличение объема циркулирующей крови при гиперосмолярной гипергидратации сопровождается развитием генерализованных отеков и транссудацией жидкости в полости тела (плевральную, перикардиальную и др.) и развитием соответствующей клинической симптоматики.
Генерализованный капиллярит
Повышенная гидрофильность тканей.
Динамическая лимфатическая недостаточность.
Нефротический отек. Он возникает вследствие преимущественного поражения тубулярного (канальцевого) аппарата почек. Для нефроза характерна длительная массивная потеря белка с мочой (протеинурия), которая ведет к гипопротеинемии и соответственно гипоонкии и, как следствие, увеличение фильтрации и снижение реабсорбции воды в капиллярах органов и тканей. В силу этого избыток воды в тканях сочетается с увеличением клубочковой фильтрацией в почках. При значительном выходе жидкости из сосудистого русла в ткани развивается гиповолемия, что служит сигналом для включения нейроэндокринных механизмов регуляции объема жидкости (волюм-рефлекс – осмо-рефлекс) и приводит к задержке в организме натрия и воды. Однако ограничение выведения воды с мочой приводит к дальнейшему потенцированию почечного отека, поскольку гипоонкия крови сохраняется (и даже может увеличиваться в связи с гемодилюцией). "Сэкономленная" почками жидкость не удерживается в крови и переходит в ткани
Патогенез почечных отеков при нефрозе.
| 1. Нарушение реабсорбции белка из-за поражения канальцев. | Альбуминурия. | Гипопротеинемия. | Онкотический механизм. |
| 2. Отставание лимфооттока от транссудации. | Динамическая лимфатическая недостаточность. | Лимфогенный механизм. | |
| 3. Уменьшение объема циркулирующей крови за счет перехода ее в ткани и полиурии. |
Выброс альдостерона. | Осмотический механизм. | |
| 4. Нарушение обмена белков, мукополисахаридов. | Повышение проницаемости капилляров. | Мембраногенный механизм. | |
Газовый ацидоз
В основе газового ацидоза лежит увеличение концентрации углекислоты и повышение ее парциального давления в крови.
Непосредственными причинами газового ацидоза могут явиться:
1) высокая концентрация СО2 во вдыхаемом воздухе;
2) недостаточность легочной вентиляции (угнетение дыхательного центра, тяжелые заболевания легких, асфиксия, неадекватное управляемое дыхание и др.);
3) недостаточность кровообращения, когда в результате резкого снижения скорости кровотока замедляется выведение углекислоты из крови.
При газовом ацидозе в крови значительно нарастает напряжение СО2, достигая 70—120 мм рт. ст. (гиперкапния). Это ведет к повышению возбудимости дыхательного центра, развивается одышка и избыток угольной кислоты в той или иной степени удаляется из организма. Данный механизм компенсации включается быстро. Быстро срабатывает также буферный механизм. Происходит нейтрализация СО2 белковым буфером плазмы крови; образующиеся при этом ионы бикарбоната частично выходят в межклеточное пространство.
Важную роль в компенсации газового ацидоза играет гемоглобин. СО2 поступает в эритроциты, в которых существенно повышается концентрация ионов Н+ и НСОз~. Избыток Н +-ионов удерживается в эритроцитах гемоглобином, а анионы НСОз- поступают в плазму в обмен на ионы хлора. Последние образуются при диссоциации NaCl, при этом освобождается Na+. Из костной ткани в обмен на ионы Н+ выходят Na и Са (может развиться остеопороз). Ионы натрия освобождаются также из фосфатного буфера — при переходе двуосновного фосфата в одноосновной, а также из белкового буфера, отдающего Na взамен на водородные ионы. Ионы натрия связываются в крови с НСОз-, при этом возрастет содержание бикарбонатов.
В процессе компенсации очень важна роль почек, в которых при высоком парциальном давлении углекислоты в крови возрастает реабсорбция Na, что обеспечивает прирост NaHCO3. Происходит усиленное выведение с мочой Н+-ионов в свободном виде или связанных с аммиаком. Для перестройки функции почек требуется время. Это поздний механизм компенсации, в полном объеме он развивается через 5—7 сут.
При установившемся газовом ацидозе, когда максимально включаются механизмы компенсации, ИО имеет положительную величину, а СБ превышают норму. При истощении компенсаторных механизмов ацидоз становится декомпенсированным, происходит сдвиг рН крови в кислую сторону.
Газовый ацидоз часто протекает на фоне кислородного голодания (из-за недостаточности функций дыхания и кровообращения), которое вследствие нарушения обменных процессов ведет к развитию метаболического ацидоза. Сочетание респираторного ацидоза с метаболическим крайне неблагоприятно и ускоряет наступление декомпенсации.
Газовый ацидоз приводит к выраженным нарушениям дыхания, кровообращения и других функций организма. Так, при гиперкапнии происходит сужение просвета бронхов, особенно выраженное при их патологии (бронхиты, бронхиальная астма и др.). Под влиянием избытка углекислоты слизистая оболочка бронхов продуцирует вязкую слизь, накопление которой способствует еще большему затруднению вентиляции. Нарастают гиперкапния и гипоксемия. Увеличение парциального давления СО2 возбуждает сосудодвигательный центр, повышается артериальное давление. При этом в результате спазма почечных артерий снижается образование мочи. Возрастает уровень катехоламинов крови, что ведет к учащению сердечных сокращений. Однако при резком избытке СО2 повышается тонус вагуса, в этом случае замедляются сокращения сердца вплоть до полной его остановки. При длительном выраженном ацидозе снижается активность адренорецепторов, что вызывает ослабление сердечной деятельности и падение артериального давления.
Негазовый ацидоз — самая частая и очень тяжелая форма нарушения кислотно-щелочного состояния крови. В основе его лежит накопление в организме нелетучих кислых продуктов (молочная, β-оксимасляная, ацетоуксусная кислоты и др.). Особенно тяжело метаболический ацидоз протекает у детей раннего возраста, так как у них ограничены щелочные резервы.
Причинами развития негазового ацидоза могут быть:
1) избыточное образование органических кислот при нарушениях обмена веществ, вызванных декомпенсированным сахарным диабетом, врожденными дефектами метаболизма, голоданием, гипоксией, общим наркозом и др.;
2) нарушение выведения из организма кислых продуктов при недостаточности функции почек (нефриты, уремия);
3) потеря организмом большого количества оснований в виде бикарбонатов с пищеварительными соками (панкреатический, кишечный) при продолжительных поносах, свищах кишечника, хирургическом удалении большой части кишечника, непроходимости кишечника. Бикарбонаты могут теряться с мочой при врожденном недоразвитии участка нефрона, ответственного за их реабсорбцию, при нефропатии беременных и др.;
4) избыточное введение минеральных кислот (отравление уксусной кислотой, длительный прием салицилатов, введение животным минеральных кислот в эксперименте и др.).
Нейтрализация избытка кислых продуктов происходит отчасти вследствие разбавления их внеклеточными жидкостями (быстро включающийся механизм). Накапливающиеся в организме кислоты связываются бикарбонатами. При этом образуется СО2 и уменьшается содержание бикарбонатов в плазме крови. Создается относительный избыток угольной кислоты, которая под влиянием фермента карбоангидразы легких разлагается на воду и СО2. Последний возбуждает дыхательный центр, вызывает гипервентиляцию и удаляется из организма. Таким образом, развивается компенсаторный газовый алкалоз. В результате гипокапнии может произойти понижение возбудимости дыхательного и сосудодвигательного центров.
В компенсацию включается и белковый буфер, который при избытке кислот ведет себя как слабое основание и нейтрализует водородные ионы. Водородные ионы переходят в эритроциты, из которых взамен в плазму выходят ионы натрия и кальция. С одной стороны, эти механизмы являются компенсаторными, так как способствуют связыванию избытка водородных ионов, с другой — они ведут к увеличению содержания в крови ионов К + , Na+, Са2+ и декальцинации костей.
Если в основе ацидоза лежит потеря бикарбонатов, то механизмы компенсации иные. В этом случае белковый буфер, реагируя с угольной кислотой, способствует увеличению содержания иона бикарбоната.
В компенсации метаболического ацидоза участвуют почки. Увеличивается выведение кислых продуктов в виде свободных кислот (β-оксимасляная, пировиноградная и др.) и аммонийных солей. Возрастает реабсорбция бикарбонатов, понижается рН мочи.
Пока в результате действия перечисленных механизмов отношение сохраняется, ацидоз остается компенсированным, хотя при этом происходят весьма существенные сдвиги (уменьшение СБ и Рсо2> отрицательная величина ИО). При истощении и недостаточности компенсаторных механизмов развивается декомпенсированный ацидоз со снижением рН. Смещение рН крови в кислую сторону приводит к тяжелым нарушениям функций организма. Нарушается работа сердца (тахикардия, экстрасистолия, в тяжелых случаях фибрилляция желудочков), снижается артериальное давление. Понижается сродство гемоглобина к кислороду, в результате чего затрудняются образование в легких оксигемоглобина и отдача гемоглобином кислорода в тканях. Этот фактор в совокупности с нарушениями сердечной деятельности ведет к развитию гипоксемии и гипоксии. При падении рН крови ниже 7,2 развивается коматозное состояние.
Среднедневные потери калия
| Локализация | Дневные потери, (мЭкв/л) |
| Почки | 5 – 10 |
| Пот | 0 – 20 |
| Кал | 40 – 120 |
Гипокалиемия – это снижение содержания калия в крови ниже 3,5 ммоль/л. Гипокалиемия всегда указывает на потерю этого иона клетками.
В качестве причин гипокалиемии указываются:
1. Уменьшение поступления калия с пищей. Редко является единственной причиной гипокалиемии, но возможно при хроническом голодании, парентеральном питании с низким содержанием К+, особенно при инфузии растворов глюкозы с инсулином.
2. Потери калия – истощение его запасов, общий или тотальный дефицит калия:
а) почечные потери калия;
б) внепочечные потери калия.
3. Повышенный транспорт калия из внеклеточного во внутриклеточное пространство.
4. Гемодилюция – снижение его концентрации в связи с введением в кровь больших количеств жидкости после дегидратации (эффект разведения).
Почечные потери калия связаны:
(1) с избыточными потерями К+ с мочой при повышенной продукции минералокортикоидов (первичный и вторичный альдостеронизм),
(2) применение диуретиков,
(3) введение в организм больших доз антибиотиков типа гентамицина, применение глюкокортикоидов, сердечных гликозидов, гипотензивных средств. При использовании таких препаратов необходимо регулярно контролировать содержание К+в плазме крови,
(4) дефицитом ионов магния (недостаток магния влечет выход из клеток калия и истощение внеклеточных запасов К+),
(5) метаболическим алкалозом и почечном канальцевом ацидозом. При метаболическом алкалозе растет почечная экскреция бикарбонат-иона, а одним из катионов, его сопровождающих, является калий. Кроме того, повышенная экскреция 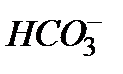 повышает канальцевую секрецию калия. На каждый секретированный ион реабсорбируется один ион К+ – формируется почечный канальцевый ацидоз.
повышает канальцевую секрецию калия. На каждый секретированный ион реабсорбируется один ион К+ – формируется почечный канальцевый ацидоз.
Внепочечные потери калия связаны:
(1) с потерей пищеварительных соков при рвоте, диарее, ворсинчатой аденоме кишечника, злоупотреблении слабительными препаратами,
(2) экссудатом из брюшной, плевральной полостей,
(3) с кожных покровов при ожогах и чрезмерном потоотделении.
Перемещение калия в клетки.
1. Увеличение захвата клетками ионов калия происходит под влиянием избытка инсулина и при стимуляции b2-адренорецепторов клеточных мембран катехоламинами (адреналин). Активирует захват калия клетками и дофамин, например, при отравлении солями бария.
2. Перераспределение калия со смещением внутрь клеток возникает при респираторных и нереспираторных алкалозах (связывают с обменом водорода на калий и натрий).
3. При усиленном синтезе гликогена и белка – гипокалиемия возникает часто после истощающих заболеваний, тяжелых операций.
Проявления гипокалиемии.
Первые клинические признаки дефицита К+ появляются при уменьшении его общего содержания в организме на 10-30%.
Гипокалиемия способствует гиперполяризации клеточных мембран, вследствие чего они теряют чувствительность к раздражителям, и возбудимость их снижается.
Это сопровождается мышечной слабостью, снижением тонуса и моторики жедудка, кишечника, мочевого пузыря. Артериальное давление падает. Тахикардия встречается реже, чем брадикардия. С гипокалиемией связывают наблюдаемые в послеоперационном периоде метеоризм и динамическую кишечную непроходимость.
В 50% случаев появляются изменения ЭКГ: удлинение интервала PQ, увеличение амплитуды зубца U, снижение сегмента ST. Однако изменения ЭКГ и недостаточность сократительной функции сердца не находятся в строгой зависимости от степени гипокалиемии. Тем не менее в генезе сердечной аритмии гипокалиемия имеет существенное значение. Легкие случаи гипокалиемии протекают бессимптомно.
Принципы коррекции гипокалиемии :
1. Пероральное восполнение дефицита калия (овощи, фрукты, курага, мясо, орехи).
2. Отмена лекарственных средств, способствующих внутриклеточному накоплению калия (например, бронхолитических средств).
3. При истощении запасов – прием препаратов калия (например, хлористого калия).
4. Компенсация метаболического алкалоза и лечение почечного канальцевого ацидоза.
Если концентрация К+ в крови не повышается под влиянием интенсивных лечебных мероприятий, следует подумать о возможном дефиците магния и провести диагностику и ликвидацию его дефицита.
Гиперкалиемия – это повышение содержания калия в крови выше 5,5 ммоль/л. Данное состояние представляет угрозу для жизни больного и гораздо серьезнее, чем гипокалиемия. Повышение содержания калия может быть обусловлено:
1. Повышенным выходом К+ из клеток;
2. Снижением экскреции К+ почками;
3.Сочетанием этих двух процессов;
4. Избыточным поступлением К+ в организм.
Повышенное поступление калия из клеток.
Выделяют несколько факторов, способствующих выбросу К+ во внеклеточное пространство:
1. Состояния массивного разрушения или распада клеток – цитолиз, травмы, ожоги;
2, Дефицит инсулина. Инсулин усиливает поглощение К+ мышцами и печенью (активация Na+, K+-АТФ-азы).
3. Ацидоз. При ацидозе отмечается повышенный обмен калия на водород через клеточную мембрану и снижение почечной экскреции калия.
4. Интоксикация препаратами наперстянки. Сердечные гликозиды ингибируют Na+, K+-АТФ-азу и сохраняют К+ внутри клетки. Компенсация этого типа гиперкалиемии осуществляется почками, постепенно повышающими экскрецию катиона.
К почечным причинам гиперкалиемии относят.
1. Почечная недостаточность с олиго- или анурией;
2. Недостатачность надпочечников – дефицит альдостерона или его неэффективность (низкий уровень секреции ренина и стимуляция ангиотензином коры надпочечников) – приводит к уменьшению экскреции калия и его задержке в организме.
3. Лекарственные средства. К ним относятся ингибиторы ангиотензинпревращающего фермента, калийсберегающие диуретики, нестероидные противовоспалительные средства. При назначении указанных средств необходимо отменить препараты калия. Гепарин обратимо, даже в малых дозах, подавляет синтез альдостерона и увеличивает концентрацию К+ .
Избыточное поступление калия наблюдается в следующих случаях:
1. Алиментарное (иногда при сочетании с калийсберегающими диуретиками);
2. При парентеральном питании;
3. Переливание длительно хранившейся крови;
4. Внутривенное введение растворов хлористого калия в высоких концентрациях.
Проявления гиперкалиемии. Выраженная гиперкалиемия (6 ммоль/л и выше) проявляется нарушением деятельности возбудимых тканей – слабостью и параличом мышц (нарушения электрогенеза и нервно-мышечной передачи), расстройствами ритма сердца.
К кардиальным проявлениям относятся брадикардия, переходящая в асистолию, замедление атрио-вентрикулярной проводимости, ведущая к атрио-вентрикулярной блокаде и фибрилляции желудочков (возможна остановка сердца в диастоле). По мере нарастания уровня калия в сыворотке меняется характер ЭКГ. Сначала появляются высокие заостренные зубцы Т, затем развивается депрессия сегмента S, расширение комплекса QRS. Скорость таких изменений непредсказуема – от начальных изменений ЭКГ до опасных нарушений проводимости и аритмий иногда проходят считанные минуты. Гиперкалиемия приводит к развитию ацидоза.
Основными принципами терапии гипергалиемии являются.
1. Улучшение процессов перехода калия в клетку. Введение инсулина и глюкозы стимулирует поглощение К+ клетками печени и мышечными волокнами скелетной мускулатуры.
2. Повышение выведения калия почками. Использование диуретиков (например, фуросемид) усиливает почечную экскрецию калия. При отсутствии благотворного действия диуретиков наиболее эффективным средством снижения содержания калия в плазме крови является диализ (гемодиализ или перитонеальный диализ).
3. Использование антагонистов калия – введение препаратов кальция, которые являются антагонистами ионов калия по влиянию на сердце.
4. Прекращение приема препаратов, содержащих калий.
68. Нарушения обмена Са2+ Виды, причины, проявления, последствия для организма.
Кальций обладает высокой биологической активностью, являясь основным структурным компонентом костей скелета и зубов, важным фактором свертывания крови. Кальций участвует в регуляции проницаемости клеточных мембран, электрогенезе нервной, мышечной и железистой ткани, процессах синаптической передачи, молекулярном механизме мышечного сокращения, контролитует ряд ферментативных процессов, оказывает влияние на процессы синтеза макроэргов. Кальций относится к трудно усвояемым элементом. Поступающие с пищей соединения кальция практически нерастворимы в воде. Под влиянием кислого содержимого желудка они частично переходят в растворимые соединения. Всасывание кальция происходит в основном в двенадцатиперстной кишке (начальной части тонкой кишки) в виде одноосновных фосфорнокислых солей и в значительной степени зависит от содержания жира, жирных кислот, витамина D.
В крови содержится 2,1-2,55 ммоль/л кальция.
Обмен кальция в организме находится под нейрогормональным контролем. Паратиреоидин (паратгормон) повышает содержание кальция в крови благодаря увеличению его всасывания в кишечнике, задержке выделения с мочой, рассасыванию костной ткани за счет растворения ее минеральных и органических компонентов. Антагонистом паратгормона является кальцитонин (тиреокальцитонин), который способен предупреждать остеопороз, подавлять распад коллагена, увеличивать экскрецию кальция с мочой. На обмен кальция оказывают также влияния соматотропный гормон, глюкокортикоиды, минералокортикоиды, тироксин, инсулин и другие гормоны.
Гиперкальциемия может возникать при избыточном поступлении солей кальция в организм (в том числе в виде лекарственных препаратов), уменьшении выведения кальция через почки при гиперпродукции паратгормона, гипервитаминозе D, разрушении костной ткани, гипотиреозе, а также иметь наследственное происхождение. Начальными проявлениями гиперкальциемии могут быть диспепсические расстройства (снижение аппетита, тошнота, рвота и т.д.), жажда и полиурия (наиболее постоянные признаки гиперкальциемии), гипотония мышц, гиперефлексия, боли в костях. Выраженные длительно текущие формы гиперкальциемии характеризуются задержкой роста у детей, кальцинозом сосудов, артериальной гипертензией, кальцификицией роговицы, грубыми нарушениями функций ЦНС.
Устранение гиперкальциемии может быть достигнуто прежде всего лечением заболевания, вызвавшего нарушения кальциевого обмена. Например, при гиперпаратиреозе единственным рациональным путем коррекции обмена кальция является хирургическое удаление гормонально активной опухоли или гиперплазированной ткани околощитовидных желез. У детей с гиперкальциемией при выявлении признаков расстройства кальциевого обмена необходимо ограничить поступление в организм витамина D. При выраженной гиперкальциемии, патологическом окостенении скелета, отложении кальция в почках, мышцах, сосудах применяют внутривенное введение динатриевой соли этилдиаминтетрауксусной кислоты, способной образовывать комплексные соединения с ионами кальция.
Гипокальциемия может возникнуть при снижении или полном прекращении продукции паратгормона (например, в результате удаления околощитовидных желез), гиповитаминозе D, уменьшении всасывания кальция в кишечнике вследствие его поражения или недостаточного выделения желчи, респираторном некомпенсированном алкалозе.
Гипокальцемия проявляется повышением нервно-мышечной возбудимости и развитии тетанических судорог, гипокоагуляцией крови, ослаблением сердечной деятельности, артериальной гипотензией. При длительной гопокальциемии возникают рахит у детей, различные трофические расстройства, в том числе, кататракта, нарушение обызвествления дентина зубов и др.
Пути устранения гипокальциемии зависят от причин ее развития и от формы возникающих расстройств. В связи с тем, что в большинстве случаев гипокальциемия является следствием ослабления или выпадения функции околощитовидных желез заместительная гормонотерапия имеет первоначальное степенное значение. В настоящее время широко применяется препарат паратиреоидного гормона – паратиреоидин. Для купирования приступов тетании у больных с выраженной гипокальциемией применяют внутривенные введения растворов хлористого кальция, глюконата или лактата кальция, а также используют препараты витамина D, дигтдротахистирол. Для устранения гипокальциемии, возникшей в результате развития алкалоза, применяют средства, направленные на коррекцию нарушения кислотно-основного состояния.
Патогенез тканевой гипоксии
Тканевая гипоксия развивается в результате ограничения способности тканей и клеток
(1) поглощать кислород вследствие нарушения процессов биологического окисления, либо
(2) в связи с ограничением эффективности сопряжения тканевого дыхания и связанного с ним окислительного фосфорилирования.
Нарушения утилизации кислорода тканями в результате угнетения биологического окисления встречается в следующих случаях:
1) при действии различных ингибиторов
2) при нарушении синтеза окислительно-восстановительных ферментов
3) при нарушении гомеостатических условий действия ферментов
4) при повреждении мембранных систем.
I. Влияние ингибиторов может происходить тремя путями:
1) неспецифическое связывание активных центров фермента. Например, активное блокирование ионом CN- конечного звена дыхательной цепочки – цитохромоксидазы при отравлении цианидами, которая обеспечивает перенос элементарных частиц на кислород. Выключение цитохромоксидазы на 93% блокирует тканевое дыхание. Аналогичная блокада возможна на уровне флавиновых ферментов, хотя их вклад в тканевое дыхание составляет лишь 7%. Алкоголь, наркотики угнетают активность дегидрогеназ. Специфическое подавление активности дыхательных ферментов вызывают также ионы сульфидов, антимицин и другие вещества.
2) Связывание функциональных групп белковой части молекулы фермента. Например, подобным действием обладают ионы тяжелых металлов, арсениты, монойодуксусная кислота и т.п. Отдельные вещества, такие, как барбитураты, антибиотики, боевые отравляющие вещества, эндо- и экзотоксины микроорганизмов могут ингибировать различные звенья дыхательной цепочки.
3) Конкурентное торможение путем блокады активного центра фермента псевдосубстратами, например, ингибирование сукцинатдегидрогеназы малоновой и другими дикарбоновыми кислотами.
II. Отклонения физико-химических параметров внутренней среды организма. Сюда следует отнести сдвиги в рН (ацидоз, алкалоз), температуры, концентрации электролитов, повышение уровня продуктов метаболизма (мочевина, билирубин, фенол, скатол и т.п.) и другие, возникающие при разнообразных заболеваниях и патологических процессах, могут существенно снижать активность ферментов биологического окисления.
III. Нарушение синтеза ферментов. Нарушение синтеза или экзогенный дефицит специфических компонентов, необходимых для образования витаминов В1 (тиамин), В2 (рибофлавин), В3 (РР, никотиновая кислота), пантотеновой кислоты и других, а также при грубых нарушениях белкового и других видов обмена (например, кахексии и т.п.).
IV. Повреждение (дезинтеграция) мембранных структур. Повреждение биологических мембран является одним из важнейших факторов, приводящих к нарушению утилизации кислорода. Чаще всего дезинтеграция мембранных структур развивается при активации перекисного свободно радикального окисления липидов (ПСОЛ), при котором органические компоненты подвергаются неферментативному окислению кислородом. Перекиси дестабилизируют мембраны митохондрий и лизосом. Активация ПСОЛ наблюдается при дефиците естественных ингибиторов свободнорадикального окисления - токоферола, рутина, убихинона, глутатиона, стероидных гормонов. Подобные изменения обусловлены многочисленными патогенными воздействиями, вызывающими повреждение клетки: высокой и низкой температурой, экзогенными ядами и эндогенными продуктами нарушенного метаболизма, инфекционно-токсическими агентами, проникающей радиацией, повышенным барометрическим давлением, гипербарической оксигенацией, свободными радикалами, а также гипоксией любого происхождения.
Еще один вид тканевой гипоксии возникает при разобщении дыхания с окислительным фосфорилированием. Потребление кислорода тканями обычно возрастает, а образование макроэргов резко ограничивается, и поэтому большая часть энергии рассеивается в виде тепла, что приводит к энергетическому обесцениванию тканевого дыхания и его относительной недостаточности. Разобщающими свойствами обладают многие вещества экзогенной и эндогенной природы: избыток ионов водорода и кальция, свободных жирных кислот, адреналина, йодсодержащих гормонов щитовидной железы (тироксина, трийодтиронина, трийодтироуксусной кислоты), а также некоторые лекарственные вещества.
Газовый состав крови в типичных случаях тканевой гипоксии характеризуется нормальными показателями кислорода в артериальной крови (раО2, объемом кислорода, сатурацией гемоглобина), значительным повышением этих параметров в венозной крови и соответственным уменьшением артерио-венозной разницы по кислороду. (При гипоксии разобщения могут возникать иные соотношения).
Обратим внимание на правомерность существования перегрузочного и субстратного типов гипоксии. Перегрузочный тип гипоксии возникает при чрезмерных нагрузках чаще всего физического характера, когда функциональные резервы систем транспорта и утилизации кислорода даже в отсутствии патологии оказываются недостаточными для обеспечения возникшей усиленной в нем потребности организма. При чрезмерной нагрузке на сердце возможно развитие коронарной недостаточности, локальная гипоксия миокарда и вторичная общая циркуляторная гипоксия. При чрезмерной мышечной работе наряду с гипоксией самой скелетной мускулатуры возникают конкурентные отношения в распределении кровотока, приводящие к ишемии других тканей и развитию распространенной гипоксии. Для гипоксии нагрузки характерны значительный кислородный долг, венозная гипоксемия и гиперкапния.
Долговременная адаптация.
Повторяющая гипоксия умеренной интенсивности способствует формированию состояния долговременной адаптации организма к гипоксии, в основе которой лежит повышение возможностей систем транспорта и утилизации кислорода:
ü стойкое увеличение диффузионной поверхности легких,
ü более совершенная корреляция вентиляции и кровотока,
ü компенсаторная гипертрофия миокарда,
ü увеличение содержание гемоглобина и эритроцитов в крови (стимуляция эритропоэза под влиянием образующихся в почках, в меньше степени печени эритропоэтинов),
ü также увеличение количества митохондрий на единицу массы клетки,
ü количественные и качественные изменения ферментов, особенно цитохромоксидазы.
При недостаточности или истощении приспособительных механизмов наступают функциональные и структурные нарушения (декомпенсация) вплоть до гибели организма. Они, как и защитно-приспособительные механизмы, развиваются в первую очередь на путях транспорта и утилизации кислорода, т.е. дыхательной, сосудистой, кровяной системах и в тканях.
Метаболические изменения раньше всего наступают в энергетическом и углеводном обмене: уменьшается содержание в клетках АТФ при одновременном увеличении концентрации продуктов его гидролиза – АДФ, АМФ и неорганического фосфата, а также креатинфосфата. Так, через несколько секунд после полного прекращения кровоснабжения мозга нейроны теряют 70% креатинфосфата, а через 40-45 сек креатинфосфат исчезает полностью. С меньшей скоростью теряется АТФ, тем не менее, дефицит АТФ в ткани мозга наступает тем скорее, чем выше функциональная активность ЦНС. Запасов макроэргов в миокарде также хватает только на несколько сокращений. Вместе с тем, значительно активируется гликолиз, вследствие чего падает содержание гиликогена и увеличивается концентрация лактата и пирувата; этому способствует также общее замедление окислительных и затруднение энергозависимых процессов ресинтеза гликогена из молочной кислоты. Несостоятельность окислительных процессов ведет за собой другие метаболические сдвиги, нарастающие по мере углубления гипоксии: нарушение обмена липидов (кетоз), белков (азотемия), электролитов (ацидоз), нейромедиаторов. По мере дальнейшего углубления гипоксии угнетается гликолиз, усиливаются процессы деструкции и распада тканей, сопровождающиеся повреждением мембран, в том числе лизосом, выходом в окружающие ткани протеолитических ферментов, вызывающих вторичную гипоксическую альтерацию, некробиоз и некроз.
Нарушения функции ЦНС начинаются в сфере высшей нервной деятельности и проявляются в расстройствах наиболее сложных аналитико-синтетических процессов.
Нарушения кровообращения выражается в тахикардии, ослаблении сократительной функции миокарда, аритмиях вплоть до фибрилляции предсердий и желудочков. Артериальное давление вначале повышается, затем прогрессивно падает вплоть до развития коллапса (паралич сосудодвигательного центра и расстройства микроциркуляции). В дыхательной системе после стадии активации (гиперпноэ) возникают диспноэтические расстройства, заканчивающиеся по мере прогрессирования гипоксии терминальным (агональным) дыханием. В крови возможно развитие гемолиза и разрушение гемоглобина.
Чувствительность различных органов и тканей к гипоксии не одинакова и зависит от нескольких факторов: интенсивности метаболизма, мощности гликолитической системы, запасов макроэргов и т.п. Например, кости, хрящи, сухожилия и мышцы мало чувствительны к гипоксии, сохраняя жизнеспособность в этих условиях в течение многих часов. Наибольшей чувствительностью к гипоксии обладают высоко дифференцированные ткани – мозг, миокард, почки, печень. Хорошо известно, что если анемизация мозга длиться более 4-6 мин, то кора головного мозга погибает, и такой пациент превращается в человека-растение. Нейроны продолговатого мозга переживают аноксию в течение 15 мин, а нейроны периферической нервной системы – в течение часа и более.
Резистентность организма к гипоксии зависит от уровня организации ЦНС, возраста (меньшая резистентность в детском возрасте, но более выраженная в эмбриональном периоде), тренировки, адаптации и акклиматизации, реактивности и функционального состояния ЦНС.
Газовый ацидоз
В основе газового ацидоза лежит увеличение концентрации углекислоты и повышение ее парциального давления в крови.
Непосредственными причинами газового ацидоза могут явиться:
1) высокая концентрация СО2 во вдыхаемом воздухе;
2) недостаточность легочной вентиляции (угнетение дыхательного центра, тяжелые заболевания легких, асфиксия, неадекватное управляемое дыхание и др.);
3) недостаточность кровообращения, когда в результате резкого снижения скорости кровотока замедляется выведение углекислоты из крови.
При газовом ацидозе в крови значительно нарастает напряжение СО2, достигая 70—120 мм рт. ст. (гиперкапния). Это ведет к повышению возбудимости дыхательного центра, развивается одышка и избыток угольной кислоты в той или иной степени удаляется из организма. Данный механизм компенсации включается быстро. Быстро срабатывает также буферный механизм. Происходит нейтрализация СО2 белковым буфером плазмы крови; образующиеся при этом ионы бикарбоната частично выходят в межклеточное пространство.
Важную роль в компенсации газового ацидоза играет гемоглобин. СО2 поступает в эритроциты, в которых существенно повышается концентрация ионов Н+ и НСОз~. Избыток Н +-ионов удерживается в эритроцитах гемоглобином, а анионы НСОз- поступают в плазму в обмен на ионы хлора. Последние образуются при диссоциации NaCl, при этом освобождается Na+. Из костной ткани в обмен на ионы Н+ выходят Na и Са (может развиться остеопороз). Ионы натрия освобождаются также из фосфатного буфера — при переходе двуосновного фосфата в одноосновной, а также из белкового буфера, отдающего Na взамен на водородные ионы. Ионы натрия связываются в крови с НСОз-, при этом возрастет содержание бикарбонатов.
В процессе компенсации очень важна роль почек, в которых при высоком парциальном давлении углекислоты в крови возрастает реабсорбция Na, что обеспечивает прирост NaHCO3. Происходит усиленное выведение с мочой Н+-ионов в свободном виде или связанных с аммиаком. Для перестройки функции почек требуется время. Это поздний механизм компенсации, в полном объеме он развивается через 5—7 сут.
При установившемся газовом ацидозе, когда максимально включаются механизмы компенсации, ИО имеет положительную величину, а СБ превышают норму. При истощении компенсаторных механизмов ацидоз становится декомпенсированным, происходит сдвиг рН крови в кислую сторону.
Газовый ацидоз часто протекает на фоне кислородного голодания (из-за недостаточности функций дыхания и кровообращения), которое вследствие нарушения обменных процессов ведет к развитию метаболического ацидоза. Сочетание респираторного ацидоза с метаболическим крайне неблагоприятно и ускоряет наступление декомпенсации.
Газовый ацидоз приводит к выраженным нарушениям дыхания, кровообращения и других функций организма. Так, при гиперкапнии происходит сужение просвета бронхов, особенно выраженное при их патологии (бронхиты, бронхиальная астма и др.). Под влиянием избытка углекислоты слизистая оболочка бронхов продуцирует вязкую слизь, накопление которой способствует еще большему затруднению вентиляции. Нарастают гиперкапния и гипоксемия. Увеличение парциального давления СО2 возбуждает сосудодвигательный центр, повышается артериальное давление. При этом в результате спазма почечных артерий снижается образование мочи. Возрастает уровень катехоламинов крови, что ведет к учащению сердечных сокращений. Однако при резком избытке СО2 повышается тонус вагуса, в этом случае замедляются сокращения сердца вплоть до полной его остановки. При длительном выраженном ацидозе снижается активность адренорецепторов, что вызывает ослабление сердечной деятельности и падение артериального давления.
Негазовый ацидоз — самая частая и очень тяжелая форма нарушения кислотно-щелочного состояния крови. В основе его лежит накопление в организме нелетучих кислых продуктов (молочная, β-оксимасляная, ацетоуксусная кислоты и др.). Особенно тяжело метаболический ацидоз протекает у детей раннего возраста, так как у них ограничены щелочные резервы.
Причинами развития негазового ацидоза могут быть:
1) избыточное образование органических кислот при нарушениях обмена веществ, вызванных декомпенсированным сахарным диабетом, врожденными дефектами метаболизма, голоданием, гипоксией, общим наркозом и др.;
2) нарушение выведения из организма кислых продуктов при недостаточности функции почек (нефриты, уремия);
3) потеря организмом большого количества оснований в виде бикарбонатов с пищеварительными соками (панкреатический, кишечный) при продолжительных поносах, свищах кишечника, хирургическом удалении большой части кишечника, непроходимости кишечника. Бикарбонаты могут теряться с мочой при врожденном недоразвитии участка нефрона, ответственного за их реабсорбцию, при нефропатии беременных и др.;
4) избыточное введение минеральных кислот (отравление уксусной кислотой, длительный прием салицилатов, введение животным минеральных кислот в эксперименте и др.).
Нейтрализация избытка кислых продуктов происходит отчасти вследствие разбавления их внеклеточными жидкостями (быстро включающийся механизм). Накапливающиеся в организме кислоты связываются бикарбонатами. При этом образуется СО2 и уменьшается содержание бикарбонатов в плазме крови. Создается относительный избыток угольной кислоты, которая под влиянием фермента карбоангидразы легких разлагается на воду и СО2. Последний возбуждает дыхательный центр, вызывает гипервентиляцию и удаляется из организма. Таким образом, развивается компенсаторный газовый алкалоз. В результате гипокапнии может произойти понижение возбудимости дыхательного и сосудодвигательного центров.
В компенсацию включается и белковый буфер, который при избытке кислот ведет себя как слабое основание и нейтрализует водородные ионы. Водородные ионы переходят в эритроциты, из которых взамен в плазму выходят ионы натрия и кальция. С одной стороны, эти механизмы являются компенсаторными, так как способствуют связыванию избытка водородных ионов, с другой — они ведут к увеличению содержания в крови ионов К + , Na+, Са2+ и декальцинации костей.
Если в основе ацидоза лежит потеря бикарбонатов, то механизмы компенсации иные. В этом случае белковый буфер, реагируя с угольной кислотой, способствует увеличению содержания иона бикарбоната.
В компенсации метаболического ацидоза участвуют почки. Увеличивается выведение кислых продуктов в виде свободных кислот (β-оксимасляная, пировиноградная и др.) и аммонийных солей. Возрастает реабсорбция бикарбонатов, понижается рН мочи.
Пока в результате действия перечисленных механизмов отношение сохраняется, ацидоз остается компенсированным, хотя при этом происходят весьма существенные сдвиги (уменьшение СБ и Рсо2> отрицательная величина ИО). При истощении и недостаточности компенсаторных механизмов развивается декомпенсированный ацидоз со снижением рН. Смещение рН крови в кислую сторону приводит к тяжелым нарушениям функций организма. Нарушается работа сердца (тахикардия, экстрасистолия, в тяжелых случаях фибрилляция желудочков), снижается артериальное давление. Понижается сродство гемоглобина к кислороду, в результате чего затрудняются образование в легких оксигемоглобина и отдача гемоглобином кислорода в тканях. Этот фактор в совокупности с нарушениями сердечной деятельности ведет к развитию гипоксемии и гипоксии. При падении рН крови ниже 7,2 развивается коматозное состояние.
Среднедневные потери калия
| Локализация | Дневные потери, (мЭкв/л) |
| Почки | 5 – 10 |
| Пот | 0 – 20 |
| Кал | 40 – 120 |
Гипокалиемия – это снижение содержания калия в крови ниже 3,5 ммоль/л. Гипокалиемия всегда указывает на потерю этого иона клетками.
В качестве причин гипокалиемии указываются:
1. Уменьшение поступления калия с пищей. Редко является единственной причиной гипокалиемии, но возможно при хроническом голодании, парентеральном питании с низким содержанием К+, особенно при инфузии растворов глюкозы с инсулином.
2. Потери калия – истощение его запасов, общий или тотальный дефицит калия:
а) почечные потери калия;
б) внепочечные потери калия.
3. Повышенный транспорт калия из внеклеточного во внутриклеточное пространство.
4. Гемодилюция – снижение его концентрации в связи с введением в кровь больших количеств жидкости после дегидратации (эффект разведения).
Почечные потери калия связаны:
(1) с избыточными потерями К+ с мочой при повышенной продукции минералокортикоидов (первичный и вторичный альдостеронизм),
(2) применение диуретиков,
(3) введение в организм больших доз антибиотиков типа гентамицина, применение глюкокортикоидов, сердечных гликозидов, гипотензивных средств. При использовании таких препаратов необходимо регулярно контролировать содержание К+в плазме крови,
(4) дефицитом ионов магния (недостаток магния влечет выход из клеток калия и истощение внеклеточных запасов К+),
(5) метаболическим алкалозом и почечном канальцевом ацидозом. При метаболическом алкалозе растет почечная экскреция бикарбонат-иона, а одним из катионов, его сопровождающих, является калий. Кроме того, повышенная экскреция повышает канальцевую секрецию калия. На каждый секретированный ион реабсорбируется один ион К+ – формируется почечный канальцевый ацидоз.
Внепочечные потери калия связаны:
(1) с потерей пищеварительных соков при рвоте, диарее, ворсинчатой аденоме кишечника, злоупотреблении слабительными препаратами,
(2) экссудатом из брюшной, плевральной полостей,
(3) с кожных покровов при ожогах и чрезмерном потоотделении.
Перемещение калия в клетки.
1. Увеличение захвата клетками ионов калия происходит под влиянием избытка инсулина и при стимуляции b2-адренорецепторов клеточных мембран катехоламинами (адреналин). Активирует захват калия клетками и дофамин, например, при отравлении солями бария.
2. Перераспределение калия со смещением внутрь клеток возникает при респираторных и нереспираторных алкалозах (связывают с обменом водорода на калий и натрий).
3. При усиленном синтезе гликогена и белка – гипокалиемия возникает часто после истощающих заболеваний, тяжелых операций.
Проявления гипокалиемии.
Первые клинические признаки дефицита К+ появляются при уменьшении его общего содержания в организме на 10-30%.
Гипокалиемия способствует гиперполяризации клеточных мембран, вследствие чего они теряют чувствительность к раздражителям, и возбудимость их снижается.
Это сопровождается мышечной слабостью, снижением тонуса и моторики жедудка, кишечника, мочевого пузыря. Артериальное давление падает. Тахикардия встречается реже, чем брадикардия. С гипокалиемией связывают наблюдаемые в послеоперационном периоде метеоризм и динамическую кишечную непроходимость.
В 50% случаев появляются изменения ЭКГ: удлинение интервала PQ, увеличение амплитуды зубца U, снижение сегмента ST. Однако изменения ЭКГ и недостаточность сократительной функции сердца не находятся в строгой зависимости от степени гипокалиемии. Тем не менее в генезе сердечной аритмии гипокалиемия имеет существенное значение. Легкие случаи гипокалиемии протекают бессимптомно.
Принципы коррекции гипокалиемии :
1. Пероральное восполнение дефицита калия (овощи, фрукты, курага, мясо, орехи).
2. Отмена лекарственных средств, способствующих внутриклеточному накоплению калия (например, бронхолитических средств).
3. При истощении запасов – прием препаратов калия (например, хлористого калия).
4. Компенсация метаболического алкалоза и лечение почечного канальцевого ацидоза.
Если концентрация К+ в крови не повышается под влиянием интенсивных лечебных мероприятий, следует подумать о возможном дефиците магния и провести диагностику и ликвидацию его дефицита.
Гиперкалиемия – это повышение содержания калия в крови выше 5,5 ммоль/л. Данное состояние представляет угрозу для жизни больного и гораздо серьезнее, чем гипокалиемия. Повышение содержания калия может быть обусловлено:
1. Повышенным выходом К+ из клеток;
2. Снижением экскреции К+ почками;
3.Сочетанием этих двух процессов;
4. Избыточным поступлением К+ в организм.
Повышенное поступление калия из клеток.
Выделяют несколько факторов, способствующих выбросу К+ во внеклеточное пространство:
1. Состояния массивного разрушения или распада клеток – цитолиз, травмы, ожоги;
2, Дефицит инсулина. Инсулин усиливает поглощение К+ мышцами и печенью (активация Na+, K+-АТФ-азы).
3. Ацидоз. При ацидозе отмечается повышенный обмен калия на водород через клеточную мембрану и снижение почечной экскреции калия.
4. Интоксикация препаратами наперстянки. Сердечные гликозиды ингибируют Na+, K+-АТФ-азу и сохраняют К+ внутри клетки. Компенсация этого типа гиперкалиемии осуществляется почками, постепенно повышающими экскрецию катиона.
К почечным причинам гиперкалиемии относят.
1. Почечная недостаточность с олиго- или анурией;
2. Недостатачность надпочечников – дефицит альдостерона или его неэффективность (низкий уровень секреции ренина и стимуляция ангиотензином коры надпочечников) – приводит к уменьшению экскреции калия и его задержке в организме.
3. Лекарственные средства. К ним относятся ингибиторы ангиотензинпревращающего фермента, калийсберегающие диуретики, нестероидные противовоспалительные средства. При назначении указанных средств необходимо отменить препараты калия. Гепарин обратимо, даже в малых дозах, подавляет синтез альдостерона и увеличивает концентрацию К+ .
Избыточное поступление калия наблюдается в следующих случаях:
1. Алиментарное (иногда при сочетании с калийсберегающими диуретиками);
2. При парентеральном питании;
3. Переливание длительно хранившейся крови;
4. Внутривенное введение растворов хлористого калия в высоких концентрациях.
Проявления гиперкалиемии. Выраженная гиперкалиемия (6 ммоль/л и выше) проявляется нарушением деятельности возбудимых тканей – слабостью и параличом мышц (нарушения электрогенеза и нервно-мышечной передачи), расстройствами ритма сердца.
К кардиальным проявлениям относятся брадикардия, переходящая в асистолию, замедление атрио-вентрикулярной проводимости, ведущая к атрио-вентрикулярной блокаде и фибрилляции желудочков (возможна остановка сердца в диастоле). По мере нарастания уровня калия в сыворотке меняется характер ЭКГ. Сначала появляются высокие заостренные зубцы Т, затем развивается депрессия сегмента S, расширение комплекса QRS. Скорость таких изменений непредсказуема – от начальных изменений ЭКГ до опасных нарушений проводимости и аритмий иногда проходят считанные минуты. Гиперкалиемия приводит к развитию ацидоза.
Основными принципами терапии гипергалиемии являются.
1. Улучшение процессов перехода калия в клетку. Введение инсулина и глюкозы стимулирует поглощение К+ клетками печени и мышечными волокнами скелетной мускулатуры.
2. Повышение выведения калия почками. Использование диуретиков (например, фуросемид) усиливает почечную экскрецию калия. При отсутствии благотворного действия диуретиков наиболее эффективным средством снижения содержания калия в плазме крови является диализ (гемодиализ или перитонеальный диализ).
3. Использование антагонистов калия – введение препаратов кальция, которые являются антагонистами ионов калия по влиянию на сердце.
4. Прекращение приема препаратов, содержащих калий.
68. Нарушения обмена Са2+ Виды, причины, проявления, последствия для организма.
Кальций обладает высокой биологической активностью, являясь основным структурным компонентом костей скелета и зубов, важным фактором свертывания крови. Кальций участвует в регуляции проницаемости клеточных мембран, электрогенезе нервной, мышечной и железистой ткани, процессах синаптической передачи, молекулярном механизме мышечного сокращения, контролитует ряд ферментативных процессов, оказывает влияние на процессы синтеза макроэргов. Кальций относится к трудно усвояемым элементом. Поступающие с пищей соединения кальция практически нерастворимы в воде. Под влиянием кислого содержимого желудка они частично переходят в растворимые соединения. Всасывание кальция происходит в основном в двенадцатиперстной кишке (начальной части тонкой кишки) в виде одноосновных фосфорнокислых солей и в значительной степени зависит от содержания жира, жирных кислот, витамина D.
В крови содержится 2,1-2,55 ммоль/л кальция.
Обмен кальция в организме находится под нейрогормональным контролем. Паратиреоидин (паратгормон) повышает содержание кальция в крови благодаря увеличению его всасывания в кишечнике, задержке выделения с мочой, рассасыванию костной ткани за счет растворения ее минеральных и органических компонентов. Антагонистом паратгормона является кальцитонин (тиреокальцитонин), который способен предупреждать остеопороз, подавлять распад коллагена, увеличивать экскрецию кальция с мочой. На обмен кальция оказывают также влияния соматотропный гормон, глюкокортикоиды, минералокортикоиды, тироксин, инсулин и другие гормоны.
Гиперкальциемия может возникать при избыточном поступлении солей кальция в организм (в том числе в виде лекарственных препаратов), уменьшении выведения кальция через почки при гиперпродукции паратгормона, гипервитаминозе D, разрушении костной ткани, гипотиреозе, а также иметь наследственное происхождение. Начальными проявлениями гиперкальциемии могут быть диспепсические расстройства (снижение аппетита, тошнота, рвота и т.д.), жажда и полиурия (наиболее постоянные признаки гиперкальциемии), гипотония мышц, гиперефлексия, боли в костях. Выраженные длительно текущие формы гиперкальциемии характеризуются задержкой роста у детей, кальцинозом сосудов, артериальной гипертензией, кальцификицией роговицы, грубыми нарушениями функций ЦНС.
Устранение гиперкальциемии может быть достигнуто прежде всего лечением заболевания, вызвавшего нарушения кальциевого обмена. Например, при гиперпаратиреозе единственным рациональным путем коррекции обмена кальция является хирургическое удаление гормонально активной опухоли или гиперплазированной ткани околощитовидных желез. У детей с гиперкальциемией при выявлении признаков расстройства кальциевого обмена необходимо ограничить поступление в организм витамина D. При выраженной гиперкальциемии, патологическом окостенении скелета, отложении кальция в почках, мышцах, сосудах применяют внутривенное введение динатриевой соли этилдиаминтетрауксусной кислоты, способной образовывать комплексные соединения с ионами кальция.
Гипокальциемия может возникнуть при снижении или полном прекращении продукции паратгормона (например, в результате удаления околощитовидных желез), гиповитаминозе D, уменьшении всасывания кальция в кишечнике вследствие его поражения или недостаточного выделения желчи, респираторном некомпенсированном алкалозе.
Гипокальцемия проявляется повышением нервно-мышечной возбудимости и развитии тетанических судорог, гипокоагуляцией крови, ослаблением сердечной деятельности, артериальной гипотензией. При длительной гопокальциемии возникают рахит у детей, различные трофические расстройства, в том числе, кататракта, нарушение обызвествления дентина зубов и др.
Пути устранения гипокальциемии зависят от причин ее развития и от формы возникающих расстройств. В связи с тем, что в большинстве случаев гипокальциемия является следствием ослабления или выпадения функции околощитовидных желез заместительная гормонотерапия имеет первоначальное степенное значение. В настоящее время широко применяется препарат паратиреоидного гормона – паратиреоидин. Для купирования приступов тетании у больных с выраженной гипокальциемией применяют внутривенные введения растворов хлористого кальция, глюконата или лактата кальция, а также используют препараты витамина D, дигтдротахистирол. Для устранения гипокальциемии, возникшей в результате развития алкалоза, применяют средства, направленные на коррекцию нарушения кислотно-основного состояния.
Патогенез тканевой гипоксии
Тканевая гипоксия развивается в результате ограничения способности тканей и клеток
(3) поглощать кислород вследствие нарушения процессов биологического окисления, либо
(4) в связи с ограничением эффективности сопряжения тканевого дыхания и связанного с ним окислительного фосфорилирования.
Нарушения утилизации кислорода тканями в результате угнетения биологического окисления встречается в следующих случаях:
5) при действии различных ингибиторов
6) при нарушении синтеза окислительно-восстановительных ферментов
7) при нарушении гомеостатических условий действия ферментов
8) при повреждении мембранных систем.
I. Влияние ингибиторов может происходить тремя путями:
4) неспецифическое связывание активных центров фермента. Например, активное блокирование ионом CN- конечного звена дыхательной цепочки – цитохромоксидазы при отравлении цианидами, которая обеспечивает перенос элементарных частиц на кислород. Выключение цитохромоксидазы на 93% блокирует тканевое дыхание. Аналогичная блокада возможна на уровне флавиновых ферментов, хотя их вклад в тканевое дыхание составляет лишь 7%. Алкоголь, наркотики угнетают активность дегидрогеназ. Специфическое подавление активности дыхательных ферментов вызывают также ионы сульфидов, антимицин и другие вещества.
5) Связывание функциональных групп белковой части молекулы фермента. Например, подобным действием обладают ионы тяжелых металлов, арсениты, монойодуксусная кислота и т.п. Отдельные вещества, такие, как барбитураты, антибиотики, боевые отравляющие вещества, эндо- и экзотоксины микроорганизмов могут ингибировать различные звенья дыхательной цепочки.
6) Конкурентное торможение путем блокады активного центра фермента псевдосубстратами, например, ингибирование сукцинатдегидрогеназы малоновой и другими дикарбоновыми кислотами.
II. Отклонения физико-химических параметров внутренней среды организма. Сюда следует отнести сдвиги в рН (ацидоз, алкалоз), температуры, концентрации электролитов, повышение уровня продуктов метаболизма (мочевина, билирубин, фенол, скатол и т.п.) и другие, возникающие при разнообразных заболеваниях и патологических процессах, могут существенно снижать активность ферментов биологического окисления.
III. Нарушение синтеза ферментов. Нарушение синтеза или экзогенный дефицит специфических компонентов, необходимых для образования витаминов В1 (тиамин), В2 (рибофлавин), В3 (РР, никотиновая кислота), пантотеновой кислоты и других, а также при грубых нарушениях белкового и других видов обмена (например, кахексии и т.п.).
IV. Повреждение (дезинтеграция) мембранных структур. Повреждение биологических мембран является одним из важнейших факторов, приводящих к нарушению утилизации кислорода. Чаще всего дезинтеграция мембранных структур развивается при активации перекисного свободно радикального окисления липидов (ПСОЛ), при котором органические компоненты подвергаются неферментативному окислению кислородом. Перекиси дестабилизируют мембраны митохондрий и лизосом. Активация ПСОЛ наблюдается при дефиците естественных ингибиторов свободнорадикального окисления - токоферола, рутина, убихинона, глутатиона, стероидных гормонов. Подобные изменения обусловлены многочисленными патогенными воздействиями, вызывающими повреждение клетки: высокой и низкой температурой, экзогенными ядами и эндогенными продуктами нарушенного метаболизма, инфекционно-токсическими агентами, проникающей радиацией, повышенным барометрическим давлением, гипербарической оксигенацией, свободными радикалами, а также гипоксией любого происхождения.
Еще один вид тканевой гипоксии возникает при разобщении дыхания с окислительным фосфорилированием. Потребление кислорода тканями обычно возрастает, а образование макроэргов резко ограничивается, и поэтому большая часть энергии рассеивается в виде тепла, что приводит к энергетическому обесцениванию тканевого дыхания и его относительной недостаточности. Разобщающими свойствами обладают многие вещества экзогенной и эндогенной природы: избыток ионов водорода и кальция, свободных жирных кислот, адреналина, йодсодержащих гормонов щитовидной железы (тироксина, трийодтиронина, трийодтироуксусной кислоты), а также некоторые лекарственные вещества.
Газовый состав крови в типичных случаях тканевой гипоксии характеризуется нормальными показателями кислорода в артериальной крови (раО2, объемом кислорода, сатурацией гемоглобина), значительным повышением этих параметров в венозной крови и соответственным уменьшением артерио-венозной разницы по кислороду. (При гипоксии разобщения могут возникать иные соотношения).
Обратим внимание на правомерность существования перегрузочного и субстратного типов гипоксии. Перегрузочный тип гипоксии возникает при чрезмерных нагрузках чаще всего физического характера, когда функциональные резервы систем транспорта и утилизации кислорода даже в отсутствии патологии оказываются недостаточными для обеспечения возникшей усиленной в нем потребности организма. При чрезмерной нагрузке на сердце возможно развитие коронарной недостаточности, локальная гипоксия миокарда и вторичная общая циркуляторная гипоксия. При чрезмерной мышечной работе наряду с гипоксией самой скелетной мускулатуры возникают конкурентные отношения в распределении кровотока, приводящие к ишемии других тканей и развитию распространенной гипоксии. Для гипоксии нагрузки характерны значительный кислородный долг, венозная гипоксемия и гиперкапния.
Долговременная адаптация.
Повторяющая гипоксия умеренной интенсивности способствует формированию состояния долговременной адаптации организма к гипоксии, в основе которой лежит повышение возможностей систем транспорта и утилизации кислорода:
ü стойкое увеличение диффузионной поверхности легких,
ü более совершенная корреляция вентиляции и кровотока,
ü компенсаторная гипертрофия миокарда,
ü увеличение содержание гемоглобина и эритроцитов в крови (стимуляция эритропоэза под влиянием образующихся в почках, в меньше степени печени эритропоэтинов),
ü также увеличение количества митохондрий на единицу массы клетки,
ü количественные и качественные изменения ферментов, особенно цитохромоксидазы.
При недостаточности или истощении приспособительных механизмов наступают функциональные и структурные нарушения (декомпенсация) вплоть до гибели организма. Они, как и защитно-приспособительные механизмы, развиваются в первую очередь на путях транспорта и утилизации кислорода, т.е. дыхательной, сосудистой, кровяной системах и в тканях.
Метаболические изменения раньше всего наступают в энергетическом и углеводном обмене: уменьшается содержание в клетках АТФ при одновременном увеличении концентрации продуктов его гидролиза – АДФ, АМФ и неорганического фосфата, а также креатинфосфата. Так, через несколько секунд после полного прекращения кровоснабжения мозга нейроны теряют 70% креатинфосфата, а через 40-45 сек креатинфосфат исчезает полностью. С меньшей скоростью теряется АТФ, тем не менее, дефицит АТФ в ткани мозга наступает тем скорее, чем выше функциональная активность ЦНС. Запасов макроэргов в миокарде также хватает только на несколько сокращений. Вместе с тем, значительно активируется гликолиз, вследствие чего падает содержание гиликогена и увеличивается концентрация лактата и пирувата; этому способствует также общее замедление окислительных и затруднение энергозависимых процессов ресинтеза гликогена из молочной кислоты. Несостоятельность окислительных процессов ведет за собой другие метаболические сдвиги, нарастающие по мере углубления гипоксии: нарушение обмена липидов (кетоз), белков (азотемия), электролитов (ацидоз), нейромедиаторов. По мере дальнейшего углубления гипоксии угнетается гликолиз, усиливаются процессы деструкции и распада тканей, сопровождающиеся повреждением мембран, в том числе лизосом, выходом в окружающие ткани протеолитических ферментов, вызывающих вторичную гипоксическую альтерацию, некробиоз и некроз.
Нарушения функции ЦНС начинаются в сфере высшей нервной деятельности и проявляются в расстройствах наиболее сложных аналитико-синтетических процессов.
Нарушения кровообращения выражается в тахикардии, ослаблении сократительной функции миокарда, аритмиях вплоть до фибрилляции предсердий и желудочков. Артериальное давление вначале повышается, затем прогрессивно падает вплоть до развития коллапса (паралич сосудодвигательного центра и расстройства микроциркуляции). В дыхательной системе после стадии активации (гиперпноэ) возникают диспноэтические расстройства, заканчивающиеся по мере прогрессирования гипоксии терминальным (агональным) дыханием. В крови возможно развитие гемолиза и разрушение гемоглобина.
Чувствительность различных органов и тканей к гипоксии не одинакова и зависит от нескольких факторов: интенсивности метаболизма, мощности гликолитической системы, запасов макроэргов и т.п. Например, кости, хрящи, сухожилия и мышцы мало чувствительны к гипоксии, сохраняя жизнеспособность в этих условиях в течение многих часов. Наибольшей чувствительностью к гипоксии обладают высоко дифференцированные ткани – мозг, миокард, почки, печень. Хорошо известно, что если анемизация мозга длиться более 4-6 мин, то кора головного мозга погибает, и такой пациент превращается в человека-растение. Нейроны продолговатого мозга переживают аноксию в течение 15 мин, а нейроны периферической нервной системы – в течение часа и более.
Резистентность организма к гипоксии зависит от уровня организации ЦНС, возраста (меньшая резистентность в детском возрасте, но более выраженная в эмбриональном периоде), тренировки, адаптации и акклиматизации, реактивности и функционального состояния ЦНС.
Вопрос 105
Стресс – общая неспецифическая реакция организма на действие раздражителей самой разной природы. Описан как набор физиологических и биохимических изменений в организме в опасной ситуации. Она протекает универсально, по общим закономерностям в ответ на разные раздражители. Селье называет стресс общим адаптационным синдромом. Он состоит из 3 фаз:
1) Реакция тревоги (alarm-реакция)– проявляется в усилении работы эндокринной системы организма. Активизируется работа гипофиза, вилочковой железы, коры надпочечников. Происходят гормональные изменения. Селье рассматривает их как полезную приспособительную реакцию организма в ответ на стресс (если стрессор очень силен для организма, то адаптации не происходит и смерть);
2) Стадия резистентности (resistance);. Заключается в восстановлении запасов тех гормонов, которые были израсходованы в состоянии тревоги. Адаптация (организм нашел возможность нормально функционировать при действии стрессора)
3) Стадия истощения. Наступает в том случае, если стрессорные условия продолжают свое воздействие на организм. Это повторная реакция тревоги, но уже без последующей стадии резистентности.
Частый стресс в полном объеме вызывает целый ряд заболеваний: язва желудка и кишечника, сердечно-сосудистые заболевания, артрит, некоторые кожные болезни, отеки.
Первоначально Селье изучал стрессорную реакцию в ответ на физические раздражители (как ОАС - общий адаптационный синдром, “синдром просто болезни”. Общий - потому, что вызывается лишь теми объектами, которые приводят к общему состоянию стресса, поскольку они воздействуют на большие области тела. Адаптационный - способствует адаптации организма к стрессовому воздействию. В качестве стрессоров при этом выступают химические, механические, температурные, психологические и др.факторы), а затем выяснилось, что и психические факторы вызывают стресс. Синдром является генерализованной реакцией всего организма и тесно связан с адаптацией. Поскольку любой агент, требующий адаптации вызывает стресс, то и любая болезнь связана с некоторым проявлением стресса (болезнь влечет за собой некоторые адаптивные реакции). Для болезней, возникающих в основном вследствие дефектов адаптации (неправильное протекание синдрома стресса), Селье предложил называние «болезни адаптации», т.к. они в меньшей степени зависят от природы патогенного фактора, чем от адаптации реакций организма на неспецифические стрессорные эффекты.
Главную роль в развитии ОАС играет состояние гиоталамо-гипофизарно-над почечниковая система (ГГН). Систему ГГН составляют следующие компоненты. Гипоталамус. В нейронах гипоталамуса вырабатывается кортиколиберин (CRF), который по аксонам достигает портальной системы гипофиза и далее в аденогипофиз (АГ). АГ секретирует кортикотропин (АКТГ), который с кровью достигает коры надпочечников и стимулирует выработку глюкокортикоидных гормонов. Глококортикоиды (кортикостерон и гидрокортизон) кровью разносятся к органам-мишеням и обуславливают развитие ОАС.
Вопрос 105
Стресс – общая неспецифическая реакция организма на действие раздражителей самой разной природы. Описан как набор физиологических и биохимических изменений в организме в опасной ситуации. Она протекает универсально, по общим закономерностям в ответ на разные раздражители. Селье называет стресс общим адаптационным синдромом. Он состоит из 3 фаз:
1) Реакция тревоги (alarm-реакция)– проявляется в усилении работы эндокринной системы организма. Активизируется работа гипофиза, вилочковой железы, коры надпочечников. Происходят гормональные изменения. Селье рассматривает их как полезную приспособительную реакцию организма в ответ на стресс (если стрессор очень силен для организма, то адаптации не происходит и смерть);
2) Стадия резистентности (resistance);. Заключается в восстановлении запасов тех гормонов, которые были израсходованы в состоянии тревоги. Адаптация (организм нашел возможность нормально функционировать при действии стрессора)
3) Стадия истощения. Наступает в том случае, если стрессорные условия продолжают свое воздействие на организм. Это повторная реакция тревоги, но уже без последующей стадии резистентности.
Частый стресс в полном объеме вызывает целый ряд заболеваний: язва желудка и кишечника, сердечно-сосудистые заболевания, артрит, некоторые кожные болезни, отеки.
Первоначально Селье изучал стрессорную реакцию в ответ на физические раздражители (как ОАС - общий адаптационный синдром, “синдром просто болезни”. Общий - потому, что вызывается лишь теми объектами, которые приводят к общему состоянию стресса, поскольку они воздействуют на большие области тела. Адаптационный - способствует адаптации организма к стрессовому воздействию. В качестве стрессоров при этом выступают химические, механические, температурные, психологические и др.факторы), а затем выяснилось, что и психические факторы вызывают стресс. Синдром является генерализованной реакцией всего организма и тесно связан с адаптацией. Поскольку любой агент, требующий адаптации вызывает стресс, то и любая болезнь связана с некоторым проявлением стресса (болезнь влечет за собой некоторые адаптивные реакции). Для болезней, возникающих в основном вследствие дефектов адаптации (неправильное протекание синдрома стресса), Селье предложил называние «болезни адаптации», т.к. они в меньшей степени зависят от природы патогенного фактора, чем от адаптации реакций организма на неспецифические стрессорные эффекты.
Главную роль в развитии ОАС играет состояние гиоталамо-гипофизарно-над почечниковая система (ГГН). Систему ГГН составляют следующие компоненты. Гипоталамус. В нейронах гипоталамуса вырабатывается кортиколиберин (CRF), который по аксонам достигает портальной системы гипофиза и далее в аденогипофиз (АГ). АГ секретирует кортикотропин (АКТГ), который с кровью достигает коры надпочечников и стимулирует выработку глюкокортикоидных гормонов. Глококортикоиды (кортикостерон и гидрокортизон) кровью разносятся к органам-мишеням и обуславливают развитие ОАС.
ПРОФИЛАКТИЧЕСКИЕ И ЛЕЧЕБНЫЕ МЕРОПРИЯТИЯ
Агенты, имеющие целью защиту интактных клеток от повреждения (профилактические) или стимуляцию адаптивных механизмов при их альтерации (лечебные), подразделяют на немедикаментозные, медикаментозные и комбинированные.
1)Немедикаментозные агенты. Немедикаментозные средства применяют, главным образом, с целью профилактики повреждения клетки. Немедикаментозные средства повышают устойчивость клеток, органов и тканей, а также организма в целом к ряду патогенных агентов.
2)Медикаментозные средства. Лекарственные средства (ЛС) применяют в основном для активации адаптивных механизмов после воздействия патогенного агента. Большинство ЛС применяется с целью этиотропной или патогенетической терапии.
3)Комбинированные воздействия. Комбинированные воздействия дают наибольший эффект, как лечебный, так и профилактический.
Дата: 2019-07-24, просмотров: 367.