Методы диспергирования заключаются в измельчении грубых частиц твердых тел до коллоидных размеров. Механизм диспергирования можно объяснить следующим способом. При деформации твердого тела на его поверхности образуются микротрещины, которые резко понижают прочность материала. Микротрещины образуются обычно в слабых местах кристаллической решетки, т.е. в местах ее дефектов, которые встречаются в среднем через 100 правильных межатомных или межмолекулярных расстояний. Слабыми местами являются границы между отдельными кристаллами или любые неоднородности. П.А.Ребиндэр, В.Д.Щукин и др. в своих работах показали, что развитие микрощелей при диспергировании происходит значительно легче в присутствии ионов электролитов и молекул поверхностно-активных веществ, которые могут адсорбироваться на поверхности микротрещин и обеспечивать расклинивающее действие. Использование ионов электролитов и ПАВ позволяет значительно повысить дисперсность измельчаемых частиц.
Впервые магнитная жидкость была получена Папелом путем размалывания в шаровой мельнице в течение 3 месяцев грубодисперсных частиц магнетита в смеси керосина и олеиновой кислоты как стабилизатора [43]. Принцип работы шаровой мельницы основан на истирании частиц материала при перекатывании стальных шаров во вращающейся мельнице. Для хорошего диспергирования объем шаров, загружаемых в мельницу, должен составлять 30-40 % от ее общего объема, а объем дисперсии не должен превышать 20 % от объема мельницы. С целью получения МЖ размол обычно проводят в жидкости-носителе. Если измельчение веществ в шаровой мельнице проводить в отсутствие дисперсионной среды, то обычно невозможно получить частицы размером меньше 600 Å. При мокром же помоле и в присутствии стабилизатора могут быть получены коллоидные растворы, с размером частиц в 100 Å.
Шаровые мельницы широко используются для получения магнитных жидкостей различного состава и имеют ряд преимуществ перед другими аппаратами для получения МЖ, а именно: измельчение проводится в замкнутом пространстве и без потерь растворителя, если он летуч, процесс можно продолжать до тех пор, пока не будет достигнута нужная степень измельчения; обслуживание машины чрезвычайно просто. Недостатком шаровых мельниц является значительное истирание шаров при работе, что приводит к нежелательному загрязнению получаемой МЖ, а также длительность и малая производительность процесса. Диспергируя в шаровых мельницах различные магнитные материалы (железо, кобальт, магнетит, ферриты и др.) в различных жидкостях-носителях (керосине, углеводородах, воде, силиконах, фторорганических и других средах), удалось получить МЖ различного состава [44-51]. Нужно отметить, что магнитные свойства магнитных жидкостей, получаемых размолом магнетита в шаровых мельницах, невысоки и с целью улучшения свойств МЖ и усовершенствования процесса получения МЖ авторы работ [52-54] предложили размалывать в жидкости-носителе более хрупкий немагнитный материал - гематит или вюстит, которые представляют собой немагнитную закись железа с дефектной структурой. При нагревании до температур эвтектической точки немагнитный вюстит превращается в магнитные материалы: магнетит и железо, по реакции
4FeO ↔ Fe3O4 + Fe
Так как реакция проходит непосредственно в жидкости-носителе и в присутствии стабилизатора, то в результате образуется устойчивая магнитная жидкость. Размол магнитных материалов осложняется магнитным притяжением частиц друг к другу, которое, суммируясь с межмолекулярным взаимодействием, приводит к укрупнению частиц и увеличению времени размола. Использование же немагнитного хрупкого вюстита позволяет сократить время размола с 1000 часов до 40 и облегчает условия их стабилизации, а полученные магнитные жидкости имеют гораздо лучшие магнитные характеристики за счет образующегося при диспропорционировании высокодисперсного железа, намагниченность которого в пять раз выше намагниченности магнетита. Основным недостатком этого метода, является потребность в высоких температурах для обеспечения условий диспропорционирования вюстита в магнетит и соответствующие ограничения по термостойкости используемых дисперсионных сред и стабилизаторов.
Попытки получить устойчивые, высокомагнитные МЖ методами диспергирования, например ультразвуком, электроплазменным измельчением, измельчением вращающимся магнитным полем или электрораспылением желаемых результатов не дали. Этими методами не удалось достичь высокой дисперсности измельчаемых материалов. Они характеризуются весьма малой производительностью, длительностью и сложностью достижения высокой дисперсности измельчаемых материалов, а получаемые МЖ - невысокой устойчивостью и слабыми магнитными свойствами.
Важную роль в получении коллоидных систем играет пептизация высокодисперсных частиц, полученных тем или иным способом, в дисперсионной среде, метод пептизации заключается в переводе в коллоидный раствор осадков, первичные частицы которых уже имеют коллоидные размеры. Пептизация может осуществляться действием электролита или поверхностно-активного вещества на осадок, промывкой осадка или химическим взаимодействием вещества с осадком, в результате чего образуется электролит, придающий устойчивость частицам дисперсной фазы. Пептизация промывкой осадка сводится к удалению из него электролита, вызвавшего коагуляцию. В результате чего оставшийся двойной электрический слой утолщается, силы отталкивания начинают преобладать над силами притяжения и отделявшиеся друг от друга мицеллы в результате броуновского движения равномерно распределяются в дисперсионной среде, т.е. образуется коллоидный раствор. Пептизация электролитами идет за счет того, что ионы электролита могут достраивать кристаллическую решетку дисперсной фазы или, адсорбируясь на поверхности, создавать двойной электрический слой, обуславливающий устойчивость коллоидной системы. Пептизация поверхностио-активными веществами обуславливается взаимным отталкиванием гибких молекул ПАВ, адсорбировавшихся на поверхности коллоидной частицы.
Задача получения устойчивых и высокомагнитых МЖ успешно решается и с использованием методов конденсации.
Методы конденсации.
Конденсационные методы основаны на соединении отдельных молекул или ионов растворенного вещества в агрегаты коллоидных размеров. Образование коллоидных систем в результате конденсации можно рассматривать как процесс кристаллизации, который протекает в две стадии:
- возникновение зародышей (центров кристаллизации);
- рост зародышей до определенного размера кристаллов. Зародыши кристаллизации, как правило, образуются в результате осаждения растворенного вещества на чужеродных мельчайших пылинках, случайно оказавшихся в системе (гетерогенная конденсация). Необходимо отметить, что вводя определенное количество чужеродных зародышей, можно получить коллоидные системы с заранее заданной дисперсностью.
Рост кристаллов происходит в результате конденсации на них ионов или молекул вещества из раствора. Важно при получении высокодисперсных систем (золей), чтобы скорость образования зародышей была велика, а скорость роста кристалликов мала, так как лишь в этом случае образуется множество кристаллов коллоидных размеров. Если же наоборот скорость образования центров кристаллизации мала, а скорость роста кристаллов велика, то все выделившееся вещество конденсируется на небольшом числе зародышей и в результате образуется небольшое количество крупных кристаллов. Важно отметить, что в первом случае будут образовываться сравнительно монодисперсные системы, а во втором - полидисперсные. Очень существенное значение для получения коллоидов имеют условия реакции, то есть температура, концентрация реагирующих веществ, скорость и порядок смешивания растворов. Используя, например, сильно разбавленные растворы, охлаждая их и осторожно перемешивая можно, в принципе, вырастить монокристалл, а используя концентрированные растворы, интенсивно перемешивая и подогревая их - частицы коллоидных размеров.
Методами конденсации высокодисперсные частицы магнетиков могут быть получены в результате химических реакций почти всех известных типов: реакций обмена, восстановления, окисления, гидролиза и т.д.
Примерами методов конденсации являются: термическое разложение карбонилов металлов, электролиз металлов из растворов их солей, соосаждение солей под действием щелочи [55-63] и другие методы.
Примером конденсационных способов может служить метод получения магнитных жидкостей на углеводородной и кремнийорганической основе с металлами, разработанный авторами работ [64-70]. Метод основан на термическом разложении карбонилов металлов непосредственно в жидкости-носителе в атмосфере инертного газа и в присутствии стабилизатора. Это позволяет защитить образующиеся высоко дисперсные частички металлов от окисления, полностью сохранить их магнитные свойства и предотвратить коагуляцию, для успешного приготовления коллоидных растворов ферромагнетиков этим методом необходимо очень точно соблюдать баланс между жидкостью-носителем, стабилизатором и металлическими частицами. Изменяя режим получения (температуру, состав и процентное соотношение исходных компонентов) можно получить коллоиды, содержащие частицы металла размером от 20 до 300Å.
Разложением пентокарбонила железа Fe(CO)5 и дикобальтоктокарбонила Cо2(CO)8 Томасом были получены устойчивые коллоиды железа и кобальта в толуоле и хлорбензоле [66-68].
Аналогично авторами [65] получены магнитные жидкости на кремний-органических соединениях с железом. При разложении карбонилов металлов образуются высокодисперсные частички чистых металлов
Fe(CO)5 ↔ Fe + 5CO
Этот метод позволяет получать магнитные жидкости на чистых металлах. Недостатком этого способа является обильное выделение токсичного оксида углерода(II), а также большая реакционная способность получаемых чистых частиц металлов, что требует особых мер предосторожности. К тому же процесс должен протекать в герметичном реакторе, в атмосфере инертного газа и при высоких температурах, что создает свои сложности.
М.А.Луниной совместно с сотрудниками усовершенствован и успешно используется для получения металлических органозолей электроконденсационный метод [40, 71]. Преимуществом ЭК-метода является возможность получения практически любых металлов в виде золей, содержащих сферические частицы со средним радиусом от 1 до 30 нм. В основе ЭК-метода лежит принцип конденсации пересыщенного пара металла, возникающего при искровом разряде высокочастотного переменного тока между грубыми частицами металла, погруженными в жидкость.
Мозговой и Блум [72] для получения коллоидной суспензии железа в толуоле также использовали электроконденсационный метод. Ими была получена довольно устойчивая коллоидная система, однако весовая концентрация железа в ней составляла лишь около 3 %.
Большой интерес представляют собой электропроводящие жидкости. В качестве жидкости-носителя используют обычно ртуть, олово, легкоплавкие металлы и их сплавы (индий, галлий и др.). Способы получения таких жидкостей основан на методе термической конденсации и электролизе [73-77].
Авторами [75] для получения коллоидных растворов гадолиния в ртути использовался термический метод конденсации. Сплав железо-гадолиний испарялся в атмосфере аргона при пониженном давлении. Образующиеся аэрозольные частицы переводились в ртуть. Концентрирование взвеси проводили испарением ртути при механическом встряхивании суспензии с целью предотвращения роста частиц. В последующих работах [76] эти авторы использовали в качестве феррофазы сплав Fe-Ni. Электролиз проводился в электролизной ванне при рН = 9,25-9,5. Катодом служила свободная поверхность ртути. Для перевода образующихся частиц во взвесь ванна подвергалась вибрации с частотой 200 Гц. Полученные магнитные жидкости имели размер частиц 30-1000Å и как указывают авторы, обладали "хорошей намагниченностью и устойчивостью" в зависимости от процентного соотношения железа и никеля. Эмерсон получил электропроводные жидкости на основе железа в ртути совмещая методы электролиза, химического замещения и термической конденсации [77]. В нескольких других работах имеются лишь краткие сообщения о полученных электропроводных жидкостей на легкоплавких металлах и их сплавах, без описания методики получения и подробных характеристик. Основная трудность на пути создания таких жидкостей состоит в отсутствии эффективных методов их стабилизации.
Обычно получение устойчивых магнитных жидкостей ведется при совокупности методов конденсации, диспергирования и пептизации.
Эти методы широко используются в технике для получения красок, пигментов, магнитных лент и т.д. Использование этих методов для производства магнитных жидкостей будет обусловлено рядом дополнительных требований, таких как:
- возможность получения ферромагнетиков с размерами частиц 50-200Å;
- возможно больший выход частиц ферромагнетиков без загрязнения и разложения,
- простота технической реализации метода,
- высокие адсорбционные свойства частиц ферромагнетика и хорошо развитая поверхность;
- хорошая устойчивость получаемых коллоидов.
Среди этих методов нужно прежде всего выделить метод получения МЖ на основе магнетита путем соосаждения его избытком щелочи из растворов солей двух- и трехвалентного железа и последующей пептизации в жидкости-носителе.
Принципиальная схема процесса представлена на рис. 5. В реакторе I соли двух- и трехвалентного железа осаждаются избытком аммиака, затем в реакторе 2 к полученному осадку магнетита при постоянном перемешивании и нагревании до 70-90°С добавляют раствор олеиновой кислоты в керосине. В реакторах 3 и 4 происходит разделение полученной магнитной жидкости от воды и растворимых солей. Для более полного отделения магнитной жидкости от воды и примесей использован магнит 4.
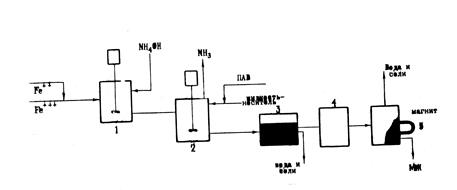
Рис.5. Технологическая схема процесса получения МЖ.
Описанный способ получения МЖ прост, производителен, позволяет получать магнетит размером 30-100Å, а магнитные жидкости - хорошей устойчивости и высокой намагниченности.
Выше рассмотрены наиболее изученные и отработанные методы получения МЖ. Но нужно отметить, что не всегда эти методы позволяют получить МЖ с широким диапазоном свойств на требуемой основе. Весьма перспективным в этом отношении является способ замены в МЖ одной жидкости-носителя другой, предложенный Р.Розенцвайгом [78] и основанный на методе пептизации. Суть этого способа состоит в том, что в МЖ вводят полярный флокулирующий агент или полимер, вызывающий флокуляцию частиц с адсорбированным на них ПАВом. В дальнейшем стабилизированные частицы выделяют из раствора и пептизируют в другой жидкости-носителе. Механизм действия флокулирующего агента приведен на рис. 6. Нa этом рисунке изображены две соседние частицы с адсорбированным на их поверхности ПАВом, взвешенные в неполярной жидкости-носителе. Когда две частицы сближаются, адсорбированные слои за счет стерического отталкивания препятствуют их укрупнению, и, естественно, и коагуляции. В силу этого частицы равномерно распределены в жидкости-носителе. При введении в систему полярного флокулирующего агента уменьшается взаимодействие ПАВ с жидкостью-носителем. Это приводит к вытеснению жидкости-носителя из зазоров между частицами и коагуляции последних с адсорбированным на них ПАВом. Скоагулировавшиеся частицы могут быть диспергированы в другой жидкости-носителе, имеющей средство к адсорбированному на них ПАВу.
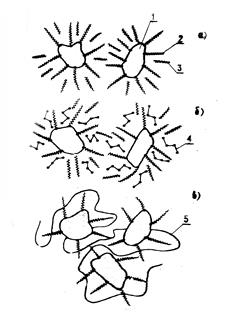
Рис.6. Механизм действия флокулирующего агента.
Флокуляция частиц полимером обусловлена большими размерами молекул полимера и их разветвленной структурой. Взаимодействуя с несколькими частицами сразу, молекула полимера обеспечивает укрупнение и коагуляцию частиц. В качестве полярного флокулирующего агента используют ацетон, этиловый спирт, этилацетат и др., а в качестве полимерных флокулянтов - полистирол, полиизобутилен, полимеры диметилсилоксана и др. Важными требованиями к полимерным флокулянтам являются их хорошая смешиваемость с жидкостью-носителем и чтобы длина молекулы в несколько раз превышала размер частиц феррофазы.
Описанный способ позволяет получать магнитные жидкости на других основах. Однако, нужно учесть, что таким образом можно производить замену сред только одного класса, например, один углеводород на другой. В дальнейшем этот способ был усовершенствован, что позволило производить замену различных по классу жидкостей-носителей [79]. Это достигается за счет подбора такого флокулирующего агента, который вызывает флокуляцию магнитных частиц без адсорбированного на них стабилизатора. Затем сфлокупировавшиеся магнитные частицы с помощью другого стабилизатора диспергируют в другой жидкости-носителе.
Замена жидкости-носителя в МЖ не только схожей с ней по свойствам, но и другими жидкостями, открывает возможность, получив предварительно МЖ наиболее простым и технологичным методом, в дальнейшем получать МЖ практически на любых основах и даже на таких, на которых прямым путем их получить очень трудно или вообще не удается. Например, можно, получив МЖ на воде, затем заменить воду углеводеродами, силиконовыми маслами, фторированными углеводородам или другими жидкостями-носителями. Кроме того, используя при повторном диспергировании другой стабилизатор, этим способом можно получить МЖ на той же основе, но с гораздо лучшими характеристиками и более широким спектром свойств.
В 80-90х гг в дипломных работах, выполненных на кафедре неорганической химии Белгосуниверситета им. В.И. Ленина дипломниками Витенчик Т.М, Омелюсик Л.С., Бурш В.В., Касперович Т.И. и др. были изучены закономерности процесса формирования пленок оксидов железа из стабильных коллоидных систем на основе магнитных оксидов железа с водной и неводной дисперсионной средой. Также было разработано несколько новых способов синтеза магнитных жидкостей. Так, например, в качестве стабилизатора были использованы олеат триэтаноламина (олеат ТЭА) и минеральные кислоты (HClO4, HNO3 и др.)
Из приведенного обзора видно, что наиболее простым и перспективным методом получения магнитных жидкостей является метод пептизации, хотя для получения МЖ с заранее заданными свойствами он требует усовершенствования.
Методика эксперимента
Синтез магнитного материала
2.1.1. Синтез магнетита
В данной работе был использован метод синтеза магнетита путем соосаждения солей двух- и трехвалентного железа избытком щелочи [80].
Готовилась смесь 25,5г FeSO4·7Н2О (марки чда) и 45г FeCl3·6Н2О, взятых в виде их 10%-ных водных растворов (т.о. отношение Fe(II):Fe(III) составляло 1,1:2, т.е. количество Fe(II) бралось в 10%-ном избытке по сравнению со стехиометрическим). Для предотвращения заметного окисления Fe(II) кислородом воздуха вода для приготовления раствора сульфата железа (II) подкислялась небольшим количеством (1-2 капли) концентрированной H2SO4.
Смесь растворов добавлялась быстро тонкой струей к 180-200мл 25%-ного раствора аммиака при интенсивном перемешивании механической мешалкой, которое продолжалось еще 20-25 мин после окончания реакции. Осаждение проводилось при рН=10.Образующийся черный осадок отмывался дистиллированной водой методом магнитной декантации до рН=8,5.
Из литературных данных известно, что целесообразно использовать 10%-е растворы солей (FeSO4·7H2O и FeCl3·6H2O) и их вливать в раствор щелочи или же проводить быструю нейтрализацию солей избытком щелочи, так как при медленном сливании разбавленных растворов образуются крупные частицы. Предотвратить образование гидрооксида железа и другие нежелательные побочные процессы можно используя предпочтительно хлорид и сульфат железа, а вместо едкого натра - водный раствор аммиака. Происходящая при этом химическая реакция может быть выражена следующим уравнением:
FeSO4·7H2O + 2FeCl3·6H2O + 8NH3·H2O ↔ Fe3O4 + 6NH4Cl + (NH4)2SO4 + 20H2O
Использование аммиака позволяет создать мягкие условия соосаждения оксидов, что благоприятствует протеканию реакции с образованием именно высоко дисперсного магнетита состава Fe3O4 или Fe2O3∙ FeO, который обладает лучшими магнитными характеристиками по сравнению с другими магнетитами, например mFe2O3∙nFeO (где n≠m), а образовавшаяся при этом соль аммония NH4Cl при нагревании легко разлагается с выделением газообразного аммиака. Ионы Сl- и растворимые соли удалялись многократной промывкой дистиллированной водой. В результате уменьшается число разноименных ионов в растворе, вызывающих коагуляцию частиц магнетита или препятствующих их пептизации в жидкости-носителе, а также снижающих впоследствии устойчивость получаемых МЖ.
Магнетит, полученный по данной методике, отличается монодисперсностью частиц, высокими магнитными свойствами, хорошей адсорбционной способностью, что является важными факторами при создании устойчивых высокомагнитных жидкостей.
2.1.2. синтез магнетита
То же что и 2.1, только соотношение Fe(II):Fe(III) =2:1
2.1.3. Синтез магнитной жидкости с водной дисперсионной средой и стабилизатором олеатом ТЭА.[81]

Рис. 7. Установка для синтеза магнитной жидкости. 1- штатив, 2 – лапка, 3 – мотор, 4 – стеклянная мешалка, 5 – пробка с отверстием, 6 – контактный термометр, 7 – стакан, 8 – водяная баня, 9 – плитка, 10 – реле, 11 – ротор.
Отдельно от магнетита, полученного по методике 2.1.1. готовился стабилизатор (смесь 9мл олеиновой кислоты и 11мл триэтаноламина). Готовый стабилизатор – олеат ТЭА – добавлялся по каплям к нагретому на водяной бане до температуры 50-60ºС осадку магнетита в воде, оставшейся после последней декантации, количество которой могло варьироваться для получения коллоидных систем с различным содержание магнетита. Нагрев и перемешивание продолжалось в течение 2х часов, затем система оставлялась для удаления пены ПАВ, образующейся в результате интенсивного перемешивания, на 0,5 -1 сутки, после чего проводилось центрифугирование для отделения фракции более крупных частиц (F = 4000g).
2.1.4. Синтез магнитной жидкости с водной дисперсионной средой и стабилизатором олеатом аммония.
Отмыв осадка, полученного аналогичным методике 2.1.1. способом, проводился до рН=9,5, после чего к дисперсии магнетита в воде (150мл), нагретой до 60ºС на водяной бане, добавлялось для стабилизации системы 20мл 25%-ного раствора аммиака, а затем 10мл олеиновой кислоты. Нагрев при 60-70ºС продолжался 1,5 часа, после чего коллоид центрифугировался и помещался в емкость из темного стекла.
2.1.5. Синтез магнитной жидкости с водной дисперсионной средой и минеральными кислотами в качестве стабилизатора
К магнетиту, приготовленному по методике 2.1.1. по каплям добавлялся очень разбавленный (0,01М) раствор азотной либо хлорной кислот до пептизации осадка, после чего проводилось центрифугирование для отделения фракции более крупных частиц (F = 4000g).
2.1.6. Синтез магнитной жидкости с деканом в качестве дисперсионной среды и стабилизатором олеиновой кислотой.[82]
Образующийся черный осадок, полученный по методике 2.1.1. после отмывания переносился в узкий стеклянный стакан. К осадку добавлялась смесь 80-100мл декана и 8-9,5 мл олеиновой кислоты, содержимое стакана перемешивалось вручную и оставлялось для расслаивания (на 1-1,5 часа) в темноте. После этого верхний органический слой (50-60мл) переносился в делительную воронку и еще раз оставлялся на 1-1,5 часа для более полного расслоения. Затем отделенный органический слой переносился в стакан емкостью 80-100мл, помещался на песчаную баню и нагревался в течение 4-5 часов при 80-90ºС при постоянном перемешивании для удаления остатка воды. Затем образец центрифугировался для удаления фракции более крупных частиц (F = 3400g) в течение 15-20 мин и помещался в емкость из темного стекла.
2.1.7. Синтез магнитной жидкости с керосином в качестве дисперсионной среды и стабилизатором олеиновой кислотой.
То же, что и 2.1.6., только вместо декана был использован керосин.
Синтез магнитного сорбента
2.2.1. Синтез намагниченного сорбента 1.
Немагнитный сорбент (активированный уголь, гранулированный активированный уголь либо ионообменный сорбент) механически смешивался с отмытым до рН=8,5 магнетитом. Далее сорбент помещался в воду и намагниченная его часть экстрагировалась при помощи постоянного магнита. Затем он высушивался при комнатной температуре и анализировался.
2.2.2. Синтез намагниченного сорбента 2.
Соосаждение смеси солей Fe(II) и Fe(III) проводилось при рН=10 в присутствии немагнитного сорбента. Отмывание магнитного сорбента проводилось при помощи магнитной декантации до рН=8,5-9. Затем намагниченная часть сорбента извлекалась при помощи постоянного магнита и высушивалась.
2.2.3. Синтез намагниченного сорбента 3.
Немагнитный сорбент (гидроксиаппатиты, активированный уголь) пропитывался различными приготовленными заранее магнитными жидкостями (с водной и неводной дисперсионной средой; отличающихся природой стабилизатора и магнитного материала (магнетит, Со)).
2.2.4. Синтез намагниченного сорбента 4.
Синтез магнетита (методика 2.1.1.) проводился в присутствии сорбента. То есть, немагнитный сорбент помещался в раствор аммиака перед соосаждением солей железа для более равномерного распределения частиц магнетита около поверхности сорбента.
2.2.5. Синтез намагниченного сорбента 5.
Соединенный с сорбентом магнетит (2.2.4.) стабилизировался по методикам 2.1.4 и 2.1.6.
2.2.6. Синтез намагниченного сорбента 6.
К полученному по методике 2.1.1. магнетиту добавлялся вместе со стабилизатором (методика 2.1.3. и 2.1.6.) сорбент.
2.2.7. Синтез намагниченного сорбента 7.






 Немагнитный легкий материал (пенопласт) пропитывался магнитной жидкостью (мет. 2.1.3.-2.1.6.) и становился магнитоуправляемым, далее наносился связующий материал (силикатный клей) и потом уже сорбент.
Немагнитный легкий материал (пенопласт) пропитывался магнитной жидкостью (мет. 2.1.3.-2.1.6.) и становился магнитоуправляемым, далее наносился связующий материал (силикатный клей) и потом уже сорбент.
|
|
|
Или:
 |  |  | |||||||
 |  | ||||||||
Методики анализа
2.3.1. Определение содержания Fe(II) при помощи количественного анализа.[81, 82]
К 1г ФМЖ прибавлялся 1мл толуола, 2мл концентрированной HCl и смесь нагревалась на кипящей водяной бане в течение 5 мин, после чего к ней добавлялось 10мл воды, 0,5 мл концентрированной H2SO4 и нагревание продолжалось еще 5 мин. Затем к смеси добавлялись 1мл концентрированной H3PO4, 4мл 5%-ного раствора MnSO4, 1мл толуола и 10мл гексана. Содержимое стакана переносилось в делительную воронку, в которой находилось 20мл воды, и после взбалтывания и отстаивания смеси нижний слой сливался в коническую колбу емкостью 100мл. В воронку повторно добавлялось 10мл воды и после встряхивания и отстаивания нижний водный слой добавлялся к полученному ранее.
Раствор в колбе титровался 0,1н раствором KMnO4 до появления розовой окраски. Параллельно проводился контрольный опыт. 1мл 0,1н раствора KMnO4 соответствует 0,005585г Fe2+.
2.3.2. Определение содержания Fe/III/ при помощи количественного анализа.[81, 82]
К 1г ФМЖ прибавлялся 1мл толуола, 2мл концентрированной HCl и смесь нагревалась на кипящей водяной бане в течение 5 мин, после чего к ней добавлялось 10мл воды, 0,5 мл концентрированной H2SO4 и нагревание продолжалось еще 5 мин. Затем к смеси добавлялись 4мл 5%-ного раствора MnSO4, 1мл толуола и 10мл гексана. Содержимое стакана переносилось в делительную воронку, в которой находилось 20мл воды, и после взбалтывания и отстаивания смеси нижний слой сливался в коническую колбу емкостью 100мл. В воронку повторно добавлялось 10мл воды и после встряхивания и отстаивания нижний водный слой добавлялся к полученному ранее. К пробе перед титрованием добавлялось 2г твердого KJ.
Раствор в колбе титровался 0,1н раствором Na2S2O3, в качестве индикатора использовался крахмал. Параллельно проводился контрольный опыт. 1мл 0,1н раствора Na2S2O3 соответствует 0,005585г Fe3+.
2.3.3. Определение содержания Fe/II/ и Fe/III/ в осадке, образующемся при соосаждении гидроксидов при помощи количественного анализа.[81, 82]
К 1г осадка, просушенного на воздухе при комнатной температуре или отжатого на фильтровальной бумаге, прибавлялось последовательно 2мл концентрированной HCl, 10мл воды, 0,5 мл концентрированной H2SO4, 4мл 5%-ного раствора MnSO4, а для определения Fe/II/ еще и 1мл концентрированной H3PO4. После растворения осадка раствор для определения Fe/II/ титровался 0,1н раствором KMnO4 до появления розовой окраски; к раствору для определения Fe/III/ добавлялось 2г твердого KJ и он оттитровывался 0,1н раствором Na2S2O3 с использованием крахмала в качестве индикатора.
Таким же образом анализировались образцы, стабилизированные минеральными кислотами.
2.3.4. Упрощенный метод определения поверхности по адсорбции воздуха.[84]
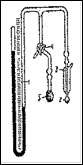
Рис.8. Прибор для определения удельной поверхности.
Прибор для определения удельной поверхности (рис.8) состоит из ртутного манометра 1 со шкалой, длина которого больше 80 см. Одна трубка манометра запаивается под вакуумом, а к другой присоединяется на шлифе ампула 2 с навеской образца. Трубка между шлифом и ампулой заключена в вакуумную рубашку 3, которая позволяет поддерживать постоянным охлаждаемый объем при погружении ампулы в жидкий азот. От трубки, соединяющей ампулу с манометром, сделан отвод с трехходовым краном 4. Вторая трубка от крана сообщается с атмосферой, а к третьей присоединена на шлифе ампула 5, содержащая несколько граммов активного угля.
Для определения удельной поверхности навеска образца помещается в ампулу, которая присоединяется к прибору. Ампула с углем соединяется с манометром и погружается в жидкий азот. После того, как весь воздух из прибора адсорбируется на угле, жидкий азот убирается и начинается десорбция газов в объем. Когда давление в манометре достигает 100—250 мм, поворотом крана ампула с углем отключается от манометра и соединяется с атмосферой. Давление газа в манометре измеряется по шкале с точностью ±0.5 мм. Ампула с образцом погружается в жидкий азот и через несколько минут определяется новое установившееся давление. Поверхность образца определяется по формуле:
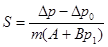 , где
, где
S – удельная поверхность,
Δp = p0 - p1
Δp0 = a0 + b0p1 (определяется по коллибровочному графику)
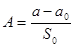
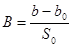
p0 и p1 – показания манометра до и после адсорбции на образце.
a, a0, b, b0, S0 – константы прибора.
Измерение адсорбции предлагаемым упрошенным методом связано с некоторыми допущениями, вносящими ошибки. Не учитывается изменение температуры жидкого азота; расчет поверхности проводится в предположении, что адсорбируется чистый азот. Между тем в воздухе неизбежно присутствует кислород. Поверхность измеряется без предварительной откачки образцов. Во многих случаях это не имеет значения, но для некоторых веществ требуется предварительный прогрев образцов для удаления адсорбированной воды.
2.3.5.Электронномикроскопическое исследование.
Препарирование образцов проводилось 2мя способами:
а) нанесения стеклянной палочкой порошка на медные опорные сеточки, расположенные на поверхности стекла и покрытые тонкой коллодиевой пленкой.
б) методом нанесения капли очень разбавленной жидкости на медные опорные сеточки, расположенные на поверхности стекла и покрытые тонкой коллодиевой пленкой.
На поверхность образцов, полученных обоими способами напылялась пленка спектрально чистого углерода толщиной 150-200 Å (вакуумный пост ВУП-4, вакуум 10-4 мм), служащая в качестве подложки при просмотре образца в электронном микроскопе.
Просмотр образцов проводился в электронном микроскопе ЭВМ – 100ЛМ. Количественная обработка результатов выполнялась по полученным ЭМ-снимкам путем определения среднего размера частиц и анализа поверхностных концентраций наблюдаемых частиц. Для количественного анализа подбирались сходные по структуре участки образцов, исследовались не менее трех участков каждого образца.
2.3.6. Рентгенографическое исследование.
Рентгенограммы образцов записывали на рентгеновском дифрактометре HZG-4A (CoKα – излучение). Расшифровка рентгенограмм велась по стандартной методике и идентифицировалась по набору межплоскостных расстояний.
2.3.7. Дериватографичеекое исследование.
Исследование проводилось на приборе "ОД-102" в воздушной атмосфере в интервале температур 25-1000°С при скорости нагрева 5 град/мин. Скорость протяжки 1 мм/мин, ДТА 1/5, ДТГ 1/15 . Дериватографическому исследованию подвергались активированный уголь, активированный уголь смешанный с магнетитом и активированный уголь пропитанный магнитной жидкостью (водн., олеат ТЭА).
Результаты и их обсуждение.
В настоящей работе представлены новые методы получения магнитных сорбентов, основанные на использовании различных магнитных жидкостей. Использование именно жидкого материала для пропитки сорбента (и придания ему тем самым магнитных свойств) выгодно отличает предложенный нами способ от описанных в литературе. Применение различных магнитных жидкостей (в отличие от магнетита определенного состава) позволяет в широких пределах варьировать свойства получаемого сорбента.
Также был проведен анализ полученных нами магнитных жидкостей и магнитных сорбентов на их основе.
3.1. Рентгенофазовое исследование.
Известно [31], что свежеосажденная смесь оксидов соответствует составу Fe3O4. Через некоторое время двухвалентное железо окисляется и Fe3O4 переходит в γ-Fe2O3. Магнетит и γ-оксид железа очень похожи по структурным характеристикам. Разница в плотности упаковки. Так, упаковка γ-оксида более плотная чем у магнетита. Разницу между ними можно обнаружить на рентгенограмме лишь в области 74-75˚ угла 2θ. У Fe3O4 межплоскостное расстояние соответствует значению 74,105˚, а у γ-Fe2O3 – 74,723˚.
Анализируя полученные нами рентгенограммы (рис.9.) можно сделать вывод, что в слабощелочной среде окисление идет быстрее, чем при рН=10. А также с течением времени магнетит стехиометрического состава переходит в nFeO·mFe2O3 где m>n, и затем уже в γ-Fe2O3.
|
| а б в | ||||||
| 80 | 75 | 70 | 65 | 60 | 55 | 50 | 2θ |
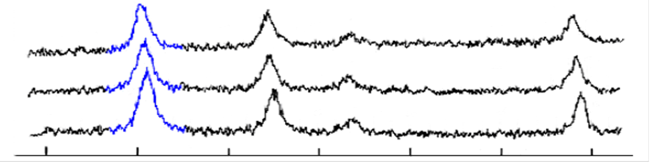
Рис. 9. Рентгенограммы образцов магнетита,
а) спустя 2 недели после синтеза, б) свежеосажденный, отмытый до рН=8,5, в) свежеосажденный, отмытый до рН=10.
|
| а б | ||||||
| 80 | 75 | 70 | 65 | 60 | 55 | 50 | 2θ |
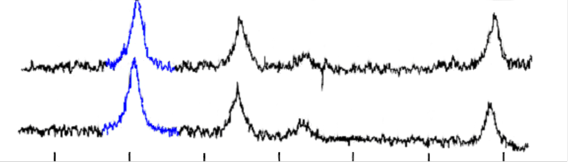
Рис. 10. Рентгенограммы образцов магнитных жидкостей в водной дисперсионной средой и смесью оксидов железа в качестве магнитного материала,
а) стабилизатор – олеат натрия, б) стабилизатор – олеат ТЭА.
Из анализа рентгенограмм (рис 10.) и данных количественного анализа следует, что в жидкостях, стабилизированных олеатом натрия, окисление двухвалентного железа идет медленнее по сравнению с жидкостями, стабилизированными олеатом ТЭА.
Что же касается концентрата магнитной жидкости на декане, то в нем, в отличие от водных МЖ и разбавленных МЖ на декане, окисление хоть и происходит, но в значительно меньшей степени.
|
| а б в | ||||||
| 80 | 75 | 70 | 65 | 60 | 55 | 50 | 2θ |
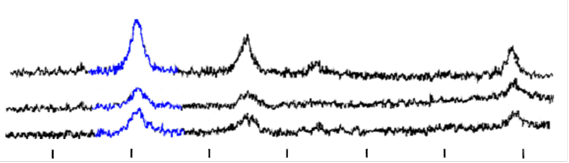
Рис. 11. Рентгенограммы а) магнетит, б) угольный магнитный сорбент, в) ионообменный магнитный сорбент
Рентгеноструктурный анализ образцов на основе магнетита (полученного путем соосаждения солей 2х- и 3х- валентного железа в аммиаке и отмытого методом магнитной декантации до рН=8,5) (рис.11) показал, что Fe2+ окисляется и со временем Fe3O4 переходит в g-Fe2O3. Это происходит примерно одинаково интенсивно в свежеосажденной смеси оксидов и магнитном сорбенте с активированным углем. В этих образцах состав магнитного материала представляет собой смесь Fe3O4 и g-Fe2O3.
При рентгеноструктурном анализе образцов магнитных сорбентов на основе магнитных жидкостей с органической и водной дисперсионной средой выяснилось, что в магнитном сорбенте на водной основе окисление происходит быстрее, чем в магнитных сорбентах, синтезированных из неводных магнитных жидкостей. А в целом, ситуация похожа на описанную выше.
3.6. Количественный анализ.
Количественный анализ проводился методами перманганато- и иодометрии. Данные представлены в табл.2 и на рис.12:
Таблица 2.
Дата: 2019-05-28, просмотров: 338.