Газы, выходящие из конденсаторов, в большинстве случаев содержат некоторое количество гексафторида урана (0,05-0,10 % об.); в них остается также некоторое количество элементного фтора и фтороводорода. Перед выбросом в атмосферу эти газы проходят еще одну обработку, вызванную необходимостью снизить содержание гексафторида урана, элементного фтора и фтороводорода до предельно допустимой концентрации.
Одним из возможных методов очистки отходящих газов является их обработка активированным углем. Процесс протекает в двух направлениях: вода, сорбированная углем, гидролизует гексафторид до нелетучего уранилфторида по реакции:
UF6 + 2H2O → UO2F2 + 4HF; (1.3.38)
углерод восстанавливает гексафторид урана до низших нелетучих фторидов UF4, U4F17, U2F9, UF5 одновременно образуя тетрафторметан по реакции:
4UF6 + C → 4UF5 + CF4 (1.3.39)
или другие фторированные углеводороды. Фтор также связывается с углеродом во фтороуглеводороды.
Этот способ очистки отходящих газов имеет некоторые недостатки: в результате фторирования углерода возможно образование взрывчатых веществ при обработке газов, содержащих обогащенный изотопом 235U гексафторид урана, высокая емкость активированного угля по урану и его отличные замедляющие свойства могут привести к взрывам.
Очистку отходящих газов можно проводить другими методами: водной или щелочной обработкой в скрубберах, поглощением гексафторида металлоорганическими соединениями, восстановлением гексафторида урана до тетрафторида урана трихлорэтиленом и другими восстановителями в газовой фазе и т. д. Однако, по-видимому, наиболее перспективные способы связаны с хемосорбцией гексафторида на тетрафториде урана.
Известно, что гексафторид урана взаимодействует с тетрафторидом, образуя промежуточные фториды. U4F17, U2F9, UF5. При температуре 204 °С парциальное давление гексафторида, находящегося в равновесии с этими фторидами, равно соответственно 8,6·10–6, 3,2·10–5 и 0,13 атм. Это дает возможность значительно снизить содержание гексафторида в газовой фазе.
Влияние различных факторов на эффективность хемосорбции гексафторида урана тетрафторидом исследовано в реакторах кипящего слоя диаметром 50 и 150 мм. Этот тип аппарата позволяет осуществить хороший контакт фаз, получить высокие коэффициенты тепло- и массопередачи и легко вести контроль температуры. Аппарат особенно удобен в том случае, когда гексафторид получают из тетрафторида урана в кипящем слое.
При проведении исследований тетрафторид урана предварительно измельчался до 50 %–325 меш (содержание фракции +80 меш было менее 5 %). Температура кипящего слоя колебалась в пределах 64-204 °С; скорость газа составляла 0,06-0,24 м/с. В этих условиях достигается хорошее псевдоожижение материала. При температуре 204 °С и исходной концентрации гексафторида урана от 0,01 до 0,35 % мас. его остаточная концентрация в газах была менее 0,0010 % мас. Емкость тетрафторида урана зависит от гранулометрического состава и колеблется в пределах 0,10-0,35 кг UF6/кг UF4. С увеличением исходной концентрации гексафторида урана в газах на входе (от 0,35 до 11 % мас.) при массовом отношении тетрафторида урана к гексафториду, равном 15 : 1, степень хемосорбции гексафторида урана падала, но во всех случаях она была не ниже 97 %. Можно полагать, что проведение процесса хемосорбции в многоступенчатом реакторе кипящего слоя даст возможность количественно поглощать гексафторид урана из газовых потоков с высокой исходной концентрацией.
Выяснено влияние свободного элементного фтора и фтороводорода на процесс хемосорбции гексафторида урана. Эффективность процесса не изменяется при концентрациях фтороводорода вплоть до 10 % мас.; таким образом, этот процесс может оказаться подходящим для регенерации гексафторида урана из фтороводорода, полученного на стадии очистки самого гексафторида. Наличие в газовой фазе до 17 % мас. свободного элементного фтора также не снижает эффективности хемосорбционного процесса; однако в этом случае при температуре 204 °С тетрафторид урана частично фторируется до промежуточных фторидов и емкость твердого вещества по гексафториду несколько снижается; около половины свободного элементного фтора поглощается тетрафторидом урана.
Проведение хемосорбционного процесса при 260 °С и выше уже нецелесообразно, так как в этом случае наблюдается значительный проскок гексафторида урана. Отмеченные закономерности дают возможность сделать важный вывод о целесообразности ступенчатого проведения хемосорбционного процесса для улавливания как гексафторида урана, так и свободного элементного фтора. Если в первом реакторе кипящего слоя поднять температуру до 240-260 °С, то свободный элементный фтор будет интенсивно поглощаться и образует промежуточные фториды урана. Проскочивший гексафторид урана, уже не содержащий свободного элементного фтора, можно поглотить во втором реакторе, температура в котором составляет 204 °С; движение газа и твердого вещества можно осуществить методом противотока.
Описанный метод очистки газов имеет то преимущество, что количественное поглощение гексафторида урана и элементного фтора сопряжено с образованием промежуточных фторидов, которые могут быть направлены на стадию получения гексафторида урана. Это уменьшит суммарный расход элементного фтора.
Для целей улавливания гексафторида урана из газов можно использовать фторид натрия (в виде гранул или таблеток).
Очистка сбросных газов при помощи фторида натрия основана на взаимодействии этого вещества с гексафторидом урана при 100 °С по реакции:
UF6 + 3NaF = 3NaF·UF6. (1.3.40)
В результате реакции образуется комплексная соль. Давление пара гексафторида урана над 3NaF·UF6 при 100 °С равно 1,5·10–3 мм. рт. ст., что соответствует давлению пара гексафторида при температуре –60 °С. При нагревании 3NaF·UF6 до 400 °С выделяется почти весь содержащийся в нем гексафторид урана. Однако процесс десорбции осложнен побочными реакциями, связанными с восстановлением урана. Для того чтобы избежать этих реакций, процесс десорбции гексафторида урана со фторида натрия проводят в токе фтора. Теоретическое содержание гексафторида урана в комплексной соли 3NaF·UF6 составляет 2,79 г UF6 на 1 г NaF; практическая емкость фторида натрия колеблется в пределах 0,6-2,1 г UF6 на 1 г NaF.
В промышленности применяется гранулированный или таблетированный фторид натрия; сорбционная система состоит из ряда периодически действующих колонн с последовательными циклами абсорбции и регенерации в токе фтора. Серьезным препятствием для эффективного улавливания гексафторида урана на гранулах или таблетках фторида натрия является фтороводород, который может присутствовать в отходящих газах. Фтороводород в температурном диапазоне 70-100 °С гораздо сильнее взаимодействует с фторидом натрия (с образованием бифторида натрия NaF·HF); равновесная концентрация гексафторида урана в газовой фазе при этом соответственно возрастает.
Применение твердых адсорбентов для улавливания относительно небольших количеств гексафторида урана из газовых потоков привлекает внимание вследствие малых капитальных и эксплуатационных затрат на цикл улавливания, хотя стоимость извлечения урана из адсорбента может быть высокой. Улавливающими материалами, которые захватывают уран в результате протекания химической реакции (кроме отмеченных выше), являются уголь и содоизвестковая смесь. Сульфат кальция, фторид кальция и оксид алюминия выделяют уран из газового потока главным образом за счет физической адсорбции.
Присутствие элементного фтора и фтороводорода оказывает большое влияние на выбор материала. Содоизвестковая смесь будет реагировать с фтороводородом и элементным фтором; уголь и сульфат кальция реагируют с элементным фтором. Фтороводород сорбируется (физическая адсорбция) на любом из вышеперечисленных материалов. Таким образом, присутствие больших количеств элементного фтора и фтороводорода в газе может значительно снизить емкость сорбента по гексафториду урана.
Для улавливания гексафторида урана из разбавленных потоков газа часто используется вода. В результате реакции образуется уранилфторид и фтороводород. Процесс проводят в вертикальных колоннах; навстречу потоку газа разбрызгивается вода. Вместо воды иногда применяют щелочные растворы (при большом содержании фтороводорода).
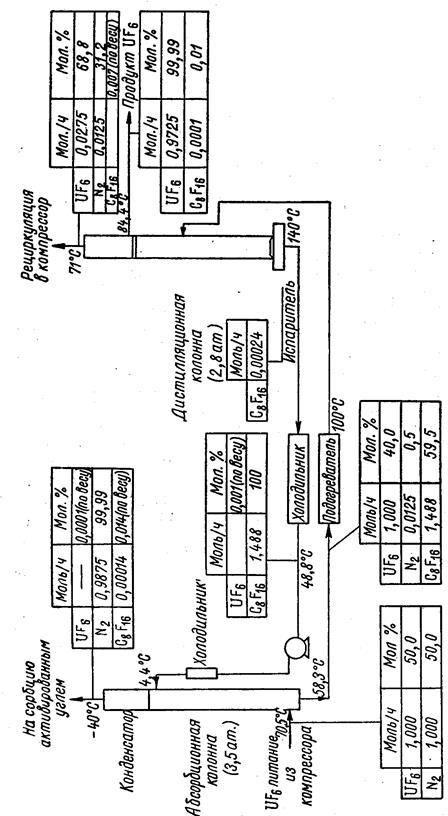
Применяется также абсорбционный способ улавливания гексафторида урана фтороуглеводородами (в частности, перфторциклогексаном) в аппаратах типа скрубберов с последующей разгонкой полученных растворов в ректификационных колоннах. Метод особенно удобен для выделения гексафторида урана из газовых потоков с высоким содержанием неконденсирующихся газов (азота и кислорода). Количественная технологическая схема процесса абсорбционного улавливания гексафторида урана перфторциклогексаном из разбавленных газовых потоков с последующей ректификацией раствора представлена на рис. 1.3.30.
Как видно из этой схемы, гексафторид урана практически нацело поглощается перфторциклогексаном; дальнейшая ректификация дает возможность сконцентрировать гексафторид урана и регенерировать растворитель.
3.2 Производство гексафторида урана методом прямого
фторирования высших оксидов урана элементным фтором
Как отмечалось ранее на российских сублиматных заводах в промышленном масштабе используют технологическую схему производства гексафторида урана методом прямого фторирования оксидов урана ядерной чистоты элементным фтором, минуя стадии восстановления высших оксидов урана водородом и гидрофторирования диоксида до тетрафторида урана. Аппаратурно-технологическая схема производства гексафторида урана прямым фторированием высших оксидов элементным фтором приведена на рис. 1.3.31.
|
Рис. 1.3.30 Количественная технологическая схема абсорбционного улавливания гексафторида урана перфторциклогексаном из разбавленных потоков с последующей ректификацией растворов |
|
1 – контейнер с исходным сырьем; 2 – противоточный реактор улавливания избытка F2; 3 – бункеры сбора твердой фазы; 4 – пламенный реактор; 5 – шнековый реактор выгрузки фторидного огарка; 6 – расширитель; 7 – контейнер сбора фторидного огарка; 8 – теплообменник; 9 – фильтр отделения пыли; 10 – бункер сбора пыли; 11 – конденсатор UF6; 12 – емкость для UF6; 13 – реактор улавливания следов фторсодержащих газов; 14 – скруббер; 15 – насос Рис. 1.3.31 Аппаратурно-технологическая схема производства гексафторида урана прямым фторированием высших оксидов анодным газом |
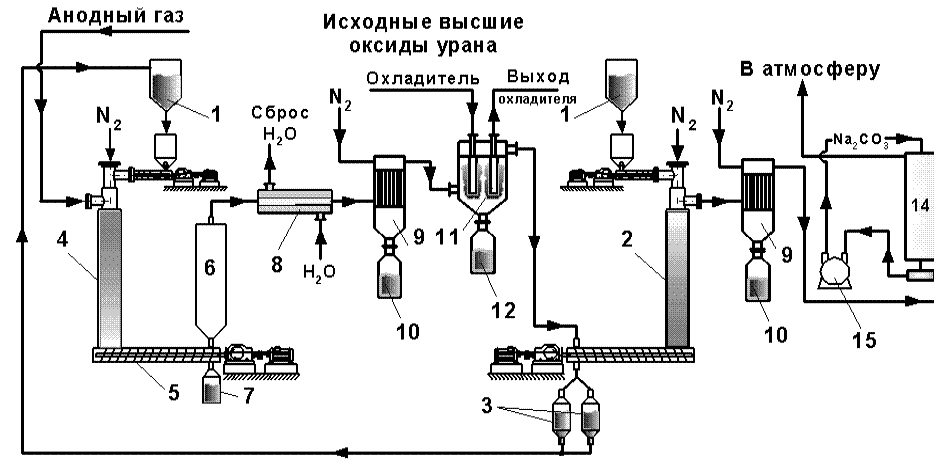
Порошкообразный твердый материал (оксиды урана) из контейнера 1 подают в аппарат шнековым питателем. Числом оборотов шнека и объемным расходом анодного газа (элементный фтор с примесью 5-7 % мас. фтороводорода) регулируется строгое соотношение реагентов газ-твердое. После смешивания эти вещества реагируют почти мгновенно по реакциям:
U3O8(тв) + 9F2(г) → 3UF6(г) + 4O2(г) + Q1, Q1 = 921,8 кДж/моль (1.3.41)
UO3(тв) + 3F2(г) → UF6(г) + 1,5O2(г) + Q2, Q2 = 888,3 кДж/моль (1.3.42)
В ходе реакций выделяется большое количество тепла (примерно в 3,5 раза больше, чем при фторировании тетрафторида урана), поэтому без предварительного нагревания оксидов урана и анодного газа в реакторе образуется пламя (факел), температура которого составляет 1500-2000 °С. Превращение оксидов урана в гексафторид происходит практически полностью в том случае, если поддерживается 10-15 %-ный относительно стехиометрии избыток фтора, а исходный порошок оксидов урана диспергируется в газовом потоке. Обеспечить однородную подачу твердого порошкообразного материала в пламенный реактор одна из самых сложных задач. Именно неоднородностью подачи твердого вещества в пламенный реактор объясняется неполное фторирование твердой фазы и образование непрореагировавшего остатка (“огарка” или “золы”). Его количество может составлять 0,5 % мас. от исходного твердого вещества.
Стенки пламенного реактора 4 охлаждаются проточной водой, подаваемой в рубашку реактора. Несмотря на высокую температуру факела, в котором протекает основная реакция сжигания оксидов урана в элементном фторе, температура стенки пламенного реактора не превышает 100-150 °С.
Технические характеристики пламенного реактора приведены в п. 3.1.3.2 настоящей главы. Общий вид реактора показан на рис. 1.3.23, а его схема представлена на рис. 1.3.24.
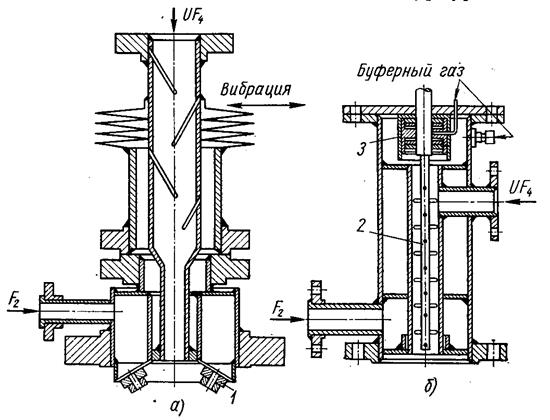
Распыление твердого порошка осуществляют с помощью вибрационного или ротационного диспергаторов, конструкции которых показаны на рис. 1.3.32, а также устройства, аналогичные используемым в пылеугольных топках.
При проектировании форсунок большой производительности необходимо учитывать их возможный разогрев и предусматривать системы охлаждения.
Для зажигания факела обычно не устанавливают специальных устройств. Реакция фторирования оксидов урана инициируется уже при комнатной температуре, поэтому предварительного нагревания исходных веществ не требуется.
Одной из важных проблем является образование на внутренних стенках пламенного реактора гарнисажа из твердых продуктов. При проведении реакции фторирования высших оксидов урана элементным фтором необходимо произвести локализацию этой реакции в объеме факела. Это достигается снижением температуры стенок пламенного реактора и введением 5-7 % избытка элементного фтора относительно стехиометрически необходимого количества. В результате на внутренних стенках реактора образуется слой гарнисажа толщиной 0,08 мм, в дальнейшем не увеличивающийся.
|
1 – форсунки; 2 – вращающийся вал; 3 – сальник Рис. 1.3.32 Вибрационный (а) и ротационный (б) диспергаторы |
Непрореагировавшие частицы высших оксидов урана попадают в горизонтальный шнековый реактор выгрузки нелетучего фторидного остатка (огарка) 5 с горизонтальной шнековой мешалкой, конструкция которого представлена на рис. 1.3.33, выгружаются в контейнер и выводятся из процесса на экстракционную переработку. Количество нелетучего фторидного остатка (огарка) не превышает 0,1-0,5 % мас. от количества оксидов урана, загружаемых в реактор на фторирование.
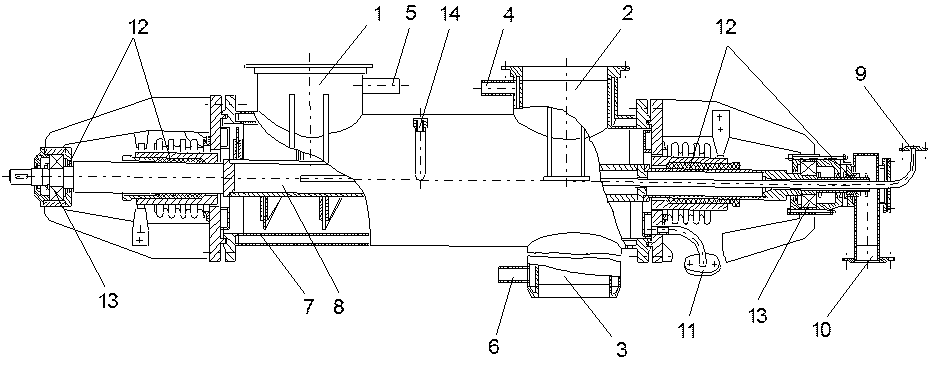
Исходный пылегазовый поток из пламенного реактора через штуцер 1 поступает в реактор выгрузки нелетучего фторидного остатка. Здесь происходит отделение основной массы твердых частиц фторидного огарка, которые шнеком 8 перемещаются по реактору 7 и удаляются через штуцер 3. Очищенный от основной массы твердых частиц пылегазовый поток выводится на дальнейшую очистку через штуцер 2. Вращающийся в реакторе 7 шнек 8 изнутри охлаждается водой, которая поступает через штуцер 9 проходит внутри вращающегося шнека 8 и сливается через штуцер 10. Для обеспечения герметичности реактора при вращении шнека предназначены сальниковые уплотнения 13. Шнек вращается при помощи вала, установленного на подшипниках 13. Для измерения температуры стенок корпуса реактора служит термопара 14. Пылегазовый поток, поступающий из пламенного реактора, имеет температуру 700-800 °С, поэтому для предотвращения коррозии реактора 7 его корпус охлаждается водой, которая подается и отводится через штуцеры 4, 5, 6 и 11.
Из реактора выгрузки нелетучего фторидного огарка 5 (рис. 1.3.31) выделенная твердая фаза собирается в контейнере 7, а очищенный от основного количества твердой фазы пылегазовый поток для дальнейшей очистки и охлаждения подается в расширитель 6, конструкция которого приведена на рис. 1.3.34.
Исходная газовая смесь поступает в расширитель через штуцер 1. В расширителе за счет резкого увеличения поперечного сечения аппарата происходит резкое падение линейной скорости газового потока. При этом некоторое количество частиц, скорость которых больше скорости газового потока выделяется из пылегазового потока и, объединяясь в агрегаты, ссыпается через штуцер 1 в реактор выгрузки фторидного огарка (рис. 1.3.33) и удаляется в контейнер 7 (рис. 1.3.31). Из расширителя частично очищенный пылегазовый поток отводится через штуцер 2. Для предотвращения коррозии корпус расширителя имеет водяную рубашку охлаждения, в которую вода подается через штуцер 3 и отводится через штуцер 4. Поэтому температура стенок расширителя не превышает 100-150 °С.
Очищенный таким способом пылегазовый поток поступает на охлаждение в теплообменник 8 (рис. 1.3.31), в котором происходит его охлаждение до комнатной температуры. Для количественного отделения пыли газовая фаза подается на металлокерамические фильтры 9, в которых фильтрация проводится с помощью порошковых спеченных фильтр-патронов. Степень очистки газа от пыли – не менее 99,99 % мас. Внутренний объем каждого фильтра разделен на 4-ре секции. Исходная пылегазовая смесь фильтруется через две секции. Одна из оставшихся секций находится на регенерации, а другая в резерве. По мере забивания фильтр-патронов одной из фильтрующих секций эта секция отключается и автоматически включается резервная секция. Отработавшая секция регенерируется и становится в резерв. Регенерация фильтр-патронов производится пропусканием сжатого элементного азота (N2) через фильтрующую поверхность в направлении обратном фильтрации пылегазового потока. Регенерация сжатым азотом производится импульсным методом в течение 0,2-0,5 с, поэтому данный процесс не оказывает значительного влияния на гидродинамику процесса фильтрации пыле-газовой смеси в работающих секциях. Отделенная пыль из фильтра сбрасывается через нижний штуцер в днище фильтра в бункер 10.
Отфильтрованный газовый поток поступает на конденсацию гексафторида урана методом вымораживания в конденсатор 11 (рис. 1.3.31).
|
1 - вход пыле-газового потока из пламенного реактора; 2 - выход пыле-газового потока в расширитель; 3 - выгрузка нелетучего фторидного остатка (огарка); 4,5 - вход и выход воды для охлаждения корпуса реактора; 6 - вода на охлаждение штуцера выгрузки фторидного огарка; 7 - корпус реактора; 8 - шнек, охлаждаемый водой; 9,10 - вход и выход воды для охлаждения шнека; 11 - выход воды для охлаждения сальникового уплотнения; 12 - сальниковые уплотнения; 13 - подшипники; 14 - термопара Рис. 1.3.33 Реактор выгрузки нелетучего фторидного остатка (огарка) |
|
1 - вход пылегазовой смеси; 2 - выход очищенной пылегазовой смеси; 3 - вход охлаждающей воды; 4 - выход охлаждающей воды Рис. 1.3.34 Расширитель |
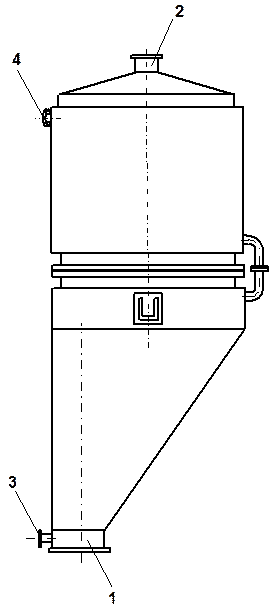
Конденсацию гексафторида урана проводят в две стадии. На 1-ой стадии в качестве охладителя используют промышленную воду с температурой 10-20 °С, а на второй – предварительно охлажденный до –40 °С раствор хлорида кальция. Выделенный из газового потока в виде твердого вещества гексафторид урана собирается в емкости 12. Основные конструкции конденсаторов UF6 представлены на рис. 1.3.26 и 1.3.27.
После конденсации газовый поток, содержащий 5-7 % избыток элементного фтора (со стадии фторирования), элементный кислород (выделившийся из оксидов урана), элементный азот (подаваемый в пламенный реактор в местах сальниковых уплотнений), фтороводород (из анодного газа) и следовые количества гексафторида урана (оставшиеся после конденсации), подается в противоточный реактор 2 (рис. 1.3.31), предназначенный для улавливания избытка элементного фтора из пламенного реактора 4. Конструкция реактора улавливания элементного фтора показана на рис. 1.3.25. Процесс улавливания проводится с помощью оксидов урана, подаваемых шнековым питателем из бункера 1 в реактор 2 в большом избытке относительно элементного фтора, содержащегося в газовой фазе. В результате в избытке оксидов урана относительно фтора протекает реакция его улавливания с образованием уранилфторида и тетрафторида урана:
U3O8(тв) + 3F2(г) = 3UO2F2(тв) + O2(г). (1.3.43)
U3O8(тв) + UF6(г) → UF4(тв) + UO2F2(тв) + 2UO3(тв). (1.3.44)
При этом достигается 99,9 %-ное улавливание элементного фтора из газовой фазы. Необходимо отметить, что лишь незначительная часть оксидов урана переходит в уранилфторид и тетрафторид урана, поэтому выгружаемая в контейнеры 3 (рис. 1.3.31) из противоточного реактора 2 твердая фаза состоит в основном из оксидов урана.
Безводный фтороводород также улавливается диоксидом урана, входящим в состав U3O8, в противоточном реакторе 2 по реакции:
UO2(тв) + 4HF(г) = UF4(тв) + 2H2O(пар). (1.3.45)
Затем полученная в процессе улавливания твердая фаза пневмотранспортом подается в контейнер 1 пламенного реактора 4.
Очищенный от основного количества фторсодержащих веществ пыле-газовый поток после фильтрации на пористом металлокерамическом фильтре 9 направляется на санитарную очистку в реактор улавливания 13 (“мокрый скруббер”), скруббер 14, через который насосом 15 противотоком газовой фазе подается 5-7 %-ный раствор соды. Улавливание фторсодержащих газов осуществляется по реакции:
F2 + Na2CO3 = 2NaF↓ + CO2 + 0,5O2, (1.3.46)
UF6 + Na2CO3 + H2O = UO2F2↓ + 2NaF↓ + 2HF + CO2↑. (1.3.47)
Нелетучий твердый остаток из контейненра 7 пламенного реактора 4 и урансодержащие растворы после санитарной очистки газовой фазы направляются на экстракционное извлечение урана.
Несмотря на то, что в качестве основных конструкционных материалов для оборудования, работающего во фторсодержащих средах, применяются никель и его сплавы, корпус пламенного реактора изготовлен из углеродистой стали. Это связано с тем, что стенки пламенного реактора с внешней стороны охлаждаются водой, поэтому их температура не превышает 100 °С. В этих условиях на внутренней поверхности стенки пламенного реактора образуется тонкая пленка из фторида железа, которая при 100 °С пассивирует поверхность углеродистой стали и препятствует проникновению элементного фтора.
Нержавеющая сталь и инконель-металл при температурах, превышающих 500 °С, совершенно неустойчивы к гексафториду урана; при температурах выше 425 °С наблюдается некоторое взаимодействие его и с монель-металлом. Исследование поведения никеля показало, что при высоких температурах скорость коррозии определяется процессом испарения пленки фторида никеля. В интервале температур 550-700 °С наблюдалась межкристаллитная коррозия, объясняемая наличием примесей в никеле. В некоторых случаях применяют плавленый алунд (Al2O3), недостатком которого, однако, является способность сорбировать некоторые количества гексафторида урана.
Критериями устойчивости металлов в среде газообразного фтора являются свойства образующихся фторидов – их летучесть и способность образовывать сплошные непроницаемые пленки на металле. Даже стойкие в среде кислорода металлы (платина, вольфрам, титан, хром) при повышенных температурах полностью разрушаются фтором. Данные по коррозионному воздействию фтора на важнейшие конструкционные материалы приведены в табл. 1.3.9.
Таблица 1.3.9
Коррозионная стойкость некоторых металлов и сплавов во фторсодержащих средах*
| Металлы и сплавы | Температура среды, °С | Скорость коррозии материала, г/(м2·ч) |
| Элементный фтор | ||
| Алюминий | 400 | 0,001 |
| Железо Амкро | 250 300 400 450 | 0,55 2,76 10,0 10,0 |
| Магний | 200-300 | 0,01 |
| Медь | 400 | 1,0 |
| Монель НМЖМц 28-2,5-1,5 | 400 500 600 | 0,149 0,573 10 |
| Никель | 400 450 500 | 0,21 0,58 1,554 |
| Фтороводород | ||
| Алюминий | 40 500 600 | 0,03** 1,5 4,53 |
| Медь | 300 500 600 | 0,98** 1,524 1,22 |
Продолжение табл.1.3.9
| Металлы и сплавы | Температура среды, °С | Скорость коррозии материала, г/(м2·ч) |
| Монель НМЖМц 28-2,5-1,5 | 43 115 | 0,022** 0,05** |
| Никель | 100 300 500 500-600 | 0,01** 0,04** 0,98 0,83 |
| Сталь 2Х13 | 500 | 8,43 |
| Сталь 12Х18Н9Т | 100 | 0,05** |
* Данные приведены для концентрации газов 90-100 %
** Данные относятся к парам плавиковой кислоты
Шкала коррозионной устойчивости металлических конструкционных материалов представлена в табл. 1.3.10.
Таблица 1.3.10
Десятибалльная шкала коррозионной устойчивости металлических1 конструкционных материалов
| Группа стойкости | Глубинный показатель коррозии, мм/год | Балл стойкости | Области применения |
| 1. Совершенно стойкие | < 0,001 | 1 | Для изготовления любых аппаратов без учета коррозионного разрушения |
| 2. Весьма стойкие | 0,001-0,005 | 2 | |
| 0,005-0,01 | 3 | ||
| 3. Стойкие | 0,01-0,05 | 4 | Для изготовления аппаратов с учетом коррозионного разрушения за время амортизации |
| 0,05-0,10 | 5 | ||
| 4. Пониженно | 0,10-0,50 | 6 | |
| 0,50-1,00 | 7 | ||
| 5. Малостойкие | 1,00-5,00 | 8 | Для изготовления неответственных, сменных деталей (патрубки, насадки и т. д.) |
| 5,00-10,00 | 9 | ||
| 6. Нестойкие | > 10,00 | 10 | Неприменимы |
| 1 Для неметаллических полимерных материалов принята трехбалльная оценка устойчивости; стоек (С), ограниченно стоек (О), нестоек (Н) | |||
Из неорганических соединений наиболее устойчивы ко фтору высшие фториды (применяют замазки на основе фторидов кальция и магния), а также, до определенных температур, алунд. Из органических соединений относительно стойки фторопласт-4 (политетрафторэтилен), в меньшей степени фторопласт-3 (политрифторхлорэтилен) и некоторые фторорганические жидкости.
Обогащение урана
3.3.1 Способы разделения изотопов урана и мощности заводов
по разделению изотопов урана в мире
Природный уран содержит около 0,7 % делящегося изотопа 235U, тогда как для работы современных LWR требуется концентрация этого изотопа 3-5 %.
Процессы обогащения урана по 235U основаны на разности масс атомов 235U и 238U и связанных с этим небольших различиях в физико-химических свойствах. На основе этих различий можно проводить разделение изотопов с помощью газовой диффузии, центрифугирования, газодинамических процессов, химического обмена, лазерными методами и т.д.
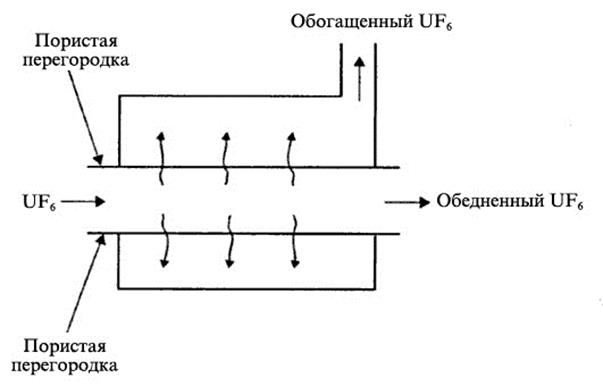
Исторически первым процессом, широко использовавшимся для промышленного обогащения урана, был процесс газовой диффузии через пористые перегородки. Разделение изотопов с помощью газодиффузионного метода основано на различии в скоростях движения молекул газообразных соединений 235U и 238U. Как известно, в смеси газов все молекулы имеют одинаковую кинетическую энергию, а для этого, в соответствии с уравнением Е=1/2∙m∙V2, молекулы с более легкой массой должны двигаться быстрее и ударяться о стенки перегородки чаще, чем тяжелые молекулы. Если перегородка будет пористой, то легкие молекулы будут проходить через нее интенсивнее, и смесь за перегородкой будет обогащена ими.
Газообразное соединение урана, пригодное для проведения процесса обогащения, должно быть летучим и химически устойчивым. Химический элемент, входящий в состав данного соединения урана, должен иметь только один изотоп и обладать низкой атомной массой. Этим требованиям удовлетворяет гексафторид урана. При комнатной температуре и атмосферном давлении он является бесцветным твердым веществом. Температура сублимации UF6 составляет 56,4 ºС.
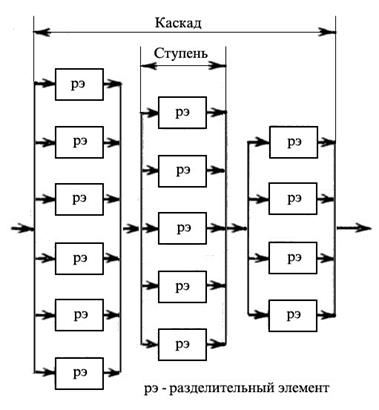
Схема диффузионного процесса через пористую перегородку показана на рис. 1.3.35. Массовые числа молекул 238UF6 и 235UF6 составляют 352 и 349, соответственно. Максимальный элементарный коэффициент разделения равен квадратному корню из отношения массовых чисел и составляет 1,0043. Однако на практике коэффициент разделения несколько меньше из-за различных технологических недостатков (в среднем около 1,001). Для достижения требуемой степени обогащения необходимо повторять процесс много раз, для чего разделительные элементы объединяются в ступени и каскады, показанные на рис. 1.3.36 и 1.3.37.
|
Рис. 1.3.35 Схема процесса газовой диффузии |
|
Рис. 1.3.36 Схема размещения разделительных элементов в ступенях и ступеней в сужающемся каскаде
|
|
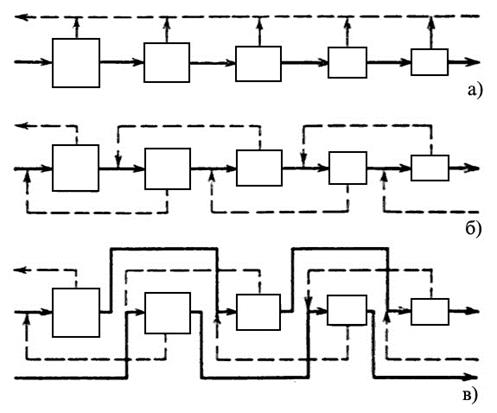
а) – простой каскад, б) – противоточный симметричный каскад, в) – противоточный несимметричный каскад с подачей потока питания через одну ступень в прямом направлении |
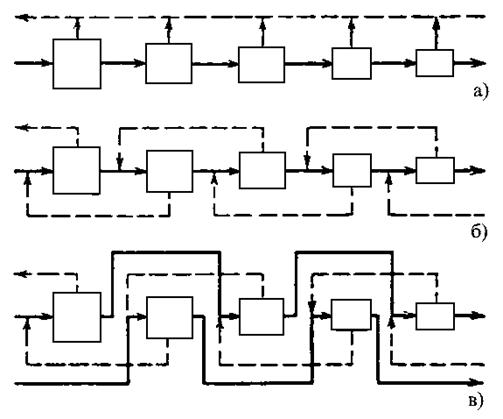
 – обедненные потоки;
– обедненные потоки; – обогащенные потоки;
– обогащенные потоки;Рис. 1.3.37 Примеры каскадов
В простом каскаде ступени соединяются последовательно, и обогащенная фракция любой ступени служит питанием для следующей ступени. При этом обедненные фракции на каждой ступени считаются отвалом и дальнейшей переработке не подлежат. В противоточном каскаде обедненная фракция, выходящая в каждой ступени, направляется на переработку в предыдущую ступень. На обогатительных заводах обычно используются противоточные каскады, так как они позволяют добиться более высокого выхода продукта.
Стандартные элементы каскада на газодиффузионных заводах включают:
1. делители, содержащие диффузионные фильтры;
2. компрессоры, устанавливаемые на каждой ступени для создания давления, необходимого для прохождения газа через фильтры;
3. теплообменники для отвода теплоты сжатия газа;
4. электромоторы, приводящие в действие компрессоры и теплообменники;
5. трубопроводы внутренних и внешних коммуникаций, клапаны и т.п.
Диффузионные фильтры, находящиеся в делителе, должны иметь малую толщину и высокую пористость, при этом поры должны быть очень мелкими. Учитывая тот факт, что фильтры должны работать продолжительное время в коррозионноактивных условиях, материалы фильтров должны быть химически инертными и обладать высокой механической прочностью. В качестве материалов для изготовления фильтров могут использоваться металлы (Au, Ag, Ni, Al, Cu), оксиды, фториды и нитриды металлов, фторопласты. Изготовление фильтров осуществляется путем протравливания тонких пластин из выбранного материала или прессования тонких порошков материала с последующим спеканием.
|
Рис. 1.3.38 Сборка трубчатых газодиффузионных фильтров |

На рис. 1.3.38 и 1.3.39 показаны сборка с фильтрами и газодиффузионная ступень в каскаде. Типичный газодиффузионный каскад, производящий низкообогащенный уран для АЭС (обогащение не выше 5 %), включает более 1000 ступеней. Для работы газодиффузионного завода требуется большое количество электроэнергии, и поэтому стоимость продукта в значительной мере зависит от ее стоимости.
|
Рис. 1.3.39 Газодиффузионная ступень в каскаде |

Газодиффузионная технология обогащения использовалась на заводах СССР, США, Франции, Великобритании, Китая и Аргентины. До 1960-1970-х гг. она была основной технологией обогащения урана в мире. Затем газовая диффузия постепенно стала вытесняться более конкурентоспособной центрифужной технологией. В настоящее время газодиффузионная технология используется только в США, Франции и Китае. В США до недавнего времени работало два газодиффузионных завода: в Портсмуте, штат Огайо, и в Падуке, штат Кентукки. Срок их службы к концу ХХ века приближался к 50 годам, и нужно было решать их дальнейшую судьбу. В результате газодиффузионный завод в Портсмуте было решено закрыть, а в здании, где в 1980-е годы испытывали центрифуги, разместить центрифужный завод. Завод в Падуке будет модернизирован и продолжит свою работу.
Во Франции действует один газодиффузионный завод, расположенный в Трикастене. Завод является рентабельным благодаря использованию дешевой электроэнергии с АЭС Трикастен и будет работать до 2010 г. Однако и на этом заводе в будущем планируется заменить газодиффузионную технологию на центрифужную. Разработка центрифуг будет осуществляться компанией Eurodif совместно с компанией Urenco.
Обогащение урана с помощью метода центрифугирования основано на эффекте разделения изотопов сильным центробежным полем во вращающемся цилиндрическом роторе. При высоких угловых скоростях в условиях вакуума более тяжелые молекулы 238UF6 двигаются к периферии, обеспечивая частичное разделение изотопов в радиальном направлении. Если создать осевой (противоточный) поток во вращательном движении газа, то степень обогащения повысится за счет добавочного обогащения в осевом направлении. Создание противотока может быть осуществлено с помощью неподвижных экранов внутри ротора, температурных градиентов и внешних насосов. В результате происходящих в центрифуге процессов тяжелая фракция с 238U опускается в нижнюю часть, а легкая с 235U поднимается вверх.
Элементарный коэффициент разделения изотопов в газовой центрифуге гораздо выше, чем в газодиффузионной ячейке. Если в газодиффузионной ячейке он, как указано выше, составляет реально около 1,001, то в газовой центрифуге – 1,09. Это позволяет снизить число ступеней разделения на центрифужном заводе примерно в 100 раз по сравнению с газодиффузионным предприятием.
|
Рис. 1.3.40 Противоточная газовая центрифуга |
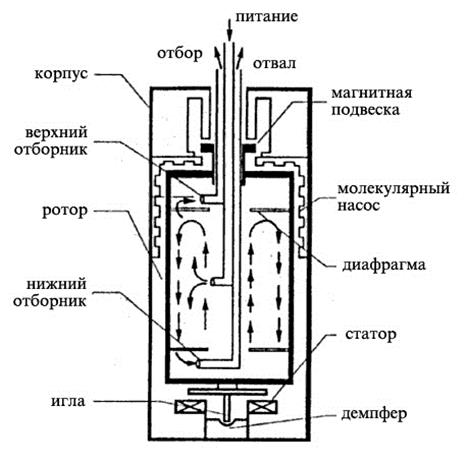
На рис. 1.3.40 изображена схема противоточной газовой центрифуги. В центре ротора установлены три концентрические трубы для подачи питания и отвода обогащенного продукта и отвала. Верхняя часть ротора удерживается в вертикальном положении с помощью магнитной подвески. Отбор продукта и отвала осуществляется вверху и внизу ротора, соответственно, с помощью газозаборников типа трубок Пито.
Производительность центрифуги в разделении изотопов пропорциональна длине ротора и скорости вращения в четвертой степени. По этой причине более предпочтительными являются центрифуги с возможно большей длиной ротора, работающие при высоких угловых скоростях. Длина ротора центрифуги ограничивается рядом практических соображений. Очень длинные роторы имеют тенденцию к изгибу, ведущему к неоднородностям газовых потоков. Кроме того, длинные роторы увеличивают стоимость оборудования для их изготовления, транспортирования роторов с заводов-изготовителей, а также стоимость производственных зданий.
Если газ с молекулярной массой М и молекулярной плотностью ρ подвергают центробежному ускорению ω2·r, то на единицу его объема действует сила Мρω2r. Здесь ω – угловая скорость и r – радиус. Градиент давления, установившийся в газе, составит:
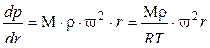 . (1.3.48)
. (1.3.48)
Для того чтобы найти отношение давлений, соответствующих радиусам 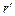 и
и 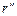 , уравнение (1.3.48) необходимо проинтегрировать. В результате получим:
, уравнение (1.3.48) необходимо проинтегрировать. В результате получим:
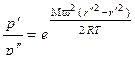 . (1.3.49)
. (1.3.49)
Полученная зависимость аналогична экспоненциальному закону изменения барометрического давления при изменении высоты.
Для смеси газов аналогичная зависимость имеет место для парциальных давлений каждого компонента. В бинарной смеси при равновесии:
– для легкого компонента
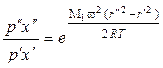 , (1.3.50)
, (1.3.50)
– для тяжелого компонента
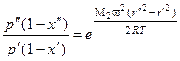 , (1.3.51)
, (1.3.51)
где М1 и М2 – молекулярные массы легкого и тяжелого компонентов; х' и х" – мольные доли легкого компонента на расстоянии от оси r' и r".
Коэффициент разделения α для аппарата вычисляют делением уравнения (1.3.51) на (1.3.50):
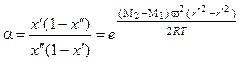 . (1.3.52)
. (1.3.52)
Необходимо отметить, что α зависит от разности молекулярных масс, а не от их отношения, в отличие от диффузионного метода разделения. В этом состоит важное преимущество метода центрифугирования, применяемого для разделения газовых смесей различных компонентов.
В качестве примера, иллюстрирующего порядок величин, рассмотрим разделение бинарной газовой смеси в центрифуге.
Пусть необходимо разделить бинарную газовую смесь изотопов гексафторидов урана 235UF6 и 238UF6).
Вычислим значения соответствующих величин:
ω = 2π·n = 2π·667 = 4190 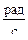 ,
,
где n – число оборотов ротора центрифуги в секунду;
М2 – М1 = = М  – М = 352 – 349 = 3
– М = 352 – 349 = 3 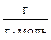 ;
;
r" = 60 мм = 6 см;
r' = 0 мм;
R = 8,314· 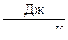 = 8,314·107
= 8,314·107 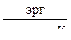 ;
;
T = 300 К.
Из уравнения (1.3.52) определим
α = 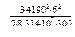 = e0,0127 = 1,01275.
= e0,0127 = 1,01275.
Эта величина примерно соответствует обогащению на одной ступени.
Материальный баланс легкого компонента на единицу высоты легкого потока при r = r' можно выразить в виде
К1 = j 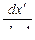 , (1.3.53)
, (1.3.53)
где dx' – вертикальный градиент концентраций легкого компонента в легком потоке, а j – поток.
Если секция обогащения каскада, производящего в секунду р– г-молей продукта с мольной долей легкого компонента хр, состоит из m включенных параллельно центрифуг, то материальный баланс аппарата описывается уравнением:
x' – x" = p·(xp – x")/j·m. (1.3.54)
Оптимальное количество центрифуг, включенных параллельно (mопт), должно соответствовать минимальной общей длине центрифуг,
т. е. величина 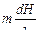 должна быть минимальной. Из этого условия найдем
должна быть минимальной. Из этого условия найдем
mопт = 2p(xp – x)/j(α – 1)·x·(1 – x). (1.3.55)
Причем
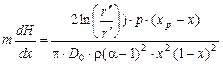 , (1.3.57)
, (1.3.57)
где D0 – коэффициент диффузии, 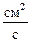 .
.
Полную длину центрифуг ΣН, необходимую в идеальном каскаде для разделения исходной газовой смеси с концентрацией хF на отдельные фракции с концентрациями xW и xp соответственно, получим интегрированием величины в уравнении (1.3.56) в пределах от x F до xp, а соответствующее выражение для секции извлечения – интегрированием в пределах от xW до хF. В результате найдем
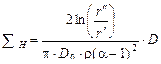 , (1.3.57)
, (1.3.57)
где D – разделительная мощность, определяемая по уравнению
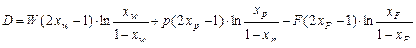 . (1.3.58)
. (1.3.58)
Разделительная мощность имеет ту же размерность, что и потоки. Она является мерой скорости, с которой каскад осуществляет разделение.
Суммарный внутренний (межступенчатый) поток в такой установке составит:
j = 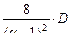 , (1.3.59)
, (1.3.59)
где разделительная мощность
D = 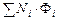 (1.3.60)
(1.3.60)
и
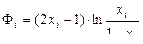 . (1.3.61)
. (1.3.61)
Функция Ф называется разделительным потенциалом. Она безразмерна, зависит только от концентрации х и симметрична относительно точки х = 0,5, в которой, как показано на рис. 1.3.41, она равна нулю.
Суммарную длину каскада центрифуг разделения можно выразить через окружную скорость u = ω·r" и отношение радиусов 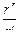 :
:
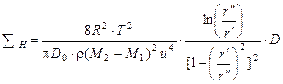 . (1.3.62)
. (1.3.62)
|
Рис. 7.14. Разделительный потенциал
Рис. 7.14. Разделительный потенциал |

|
Рис. 1.3.41 Разделительный потенциал |

Из уравнения (1.3.62) видно, что размеры установки обратно пропорциональны четвертой степени окружной скорости ротора центрифуги. В результате имеем, что 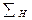 не зависит от абсолютной величины радиусов, но существует оптимальное значение отношения
не зависит от абсолютной величины радиусов, но существует оптимальное значение отношения 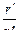 , при котором второй множитель уравнения минимален. Это имеет место при
, при котором второй множитель уравнения минимален. Это имеет место при 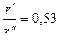 .
.
Центрифуги могут быть надкритическими и подкритическими в зависимости от того, работают ли они выше или ниже резонансной частоты, при которой происходят явления вибрации ротора. Во избежание разрушения центрифуги должны вращаться с частотами, далекими от резонансных, и должны иметь пусковые и тормозные устройства, позволяющие быстро проходить резонансные частоты. Надкритические центрифуги являются более перспективными с технических и экономических позиций.
Для сооружения высокоскоростных центрифуг необходимы особо прочные материалы. Для роторов, выполненных из алюминия или нержавеющей стали, максимальные угловые скорости составляют около 400 м/с. Современные композитные материалы позволяют увеличивать скорость до 700 м/с и выше, что значительно увеличивает эффективность разделения изотопов. В настоящее время разработано и применяется в промышленности большое количество центрифуг, различающихся по диаметру и длине ротора, скоростям вращения и другим инженерным особенностям.
Основное преимущество центрифужного метода заключается в малой энергоемкости, в несколько десятков раз меньшей, чем энергоемкость газодиффузионного метода. Кроме того, для заданной степени обогащения урана требуется существенно меньшее число ступеней по сравнению с газовой диффузией. Поэтому центрифужная технология оказалась более выгодной экономически и начала постепенно вытеснять газодиффузионную технологию.
Центрифужные обогатительные заводы работают в России, Великобритании, Нидерландах, Германии, Японии, Китае. Небольшая установка имеется в Пакистане. Наибольшими мощностями по центрифужному обогащению располагает Россия, которая эксплуатирует четыре завода суммарной производительностью 15 млн. кг ЕРР/год. Центрифужная технология, как уже упоминалось выше, будет внедрена на французском заводе в Трикастене. В США в Портсмуте в 2005 г. планируется ввести в строй первый в стране обогатительный завод на базе центрифужной технологии. На заводе будет установлено 240 центрифуг. Центрифуги разрабатывались более 20 лет в Министерстве Энергетики, их испытания предполагают проводить в Ок-Ридже. Центрифужный завод мощностью 0,1 млн. кг ЕРР/год строится в Бразилии в Резенде.
Таким образом, мировая обогатительная промышленность базируется на двух основных методах: газодиффузионном и центрифужном. Работа над остальными методами либо законсервирована, либо пока не доведена до промышленной реализации. Среди них следует упомянуть следующие методы.
1. Электромагнитный (масс-спектрографический) метод. Разделение основано на различном отклонении ионов разных изотопов в электромагнитном поле. Метод разрабатывался в СССР и США, однако из-за малой производительности, высокой энергоемкости и больших эксплуатационных затрат данная технология не нашла промышленного применения в обогащении урана.
2. Тепловая диффузия. При создании разности температур на противоположных стенках ячейки более легкий 235U диффундирует в сторону горячей зоны быстрее, чем тяжелый 238U. С использованием этой технологии в США был построен опытный завод, который проработал с 1944 по 1945 гг.
3. Химический обмен. Метод базируется на реакциях обмена между химическими соединениями, находящимися в двух несмешивающихся фазах. В результате одна из фаз обогащается 235U. Этот метод лег в основу процесса Chemex (Франция), где несмешивающими фазами являются вода и органическая жидкость, и процесса Asahi (Япония), в котором одна фаза – жидкость, а вторая – твердый адсорбент. Процессы химического обмена широко используются в ядерной промышленности для разделения легких изотопов, например, для получения 10В и тяжелой воды.
4. Газодинамические процессы. Основаны на подаче газового потока с большой скоростью на криволинейную стенку, в результате чего легкие и тяжелые изотопы отклоняются стенкой по-разному. Технологическим газом служит UF6 (~4 %) в водороде или гелии. Разрабатывалось два вида газодинамических систем: с разделительным соплом Бекера (Германия) и с вихревой трубкой (ЮАР). В ЮАР с 1987 по 1995 гг. действовала опытно-промышленная установка производительностью 0,3 млн. кг ЕРР/год.
5. Лазерные методы. Базируются на различии в спектрах поглощения атомов 235U и 238U или молекул, содержащих эти атомы. Лазерное излучение определенной частоты поглощается атомами или молекулами только одного изотопа, переводя их в возбужденное состояние. Для выделения возбужденных атомов или молекул из смеси можно использовать процессы их последующей ионизации, диссоциации или реакции с подходящим химическим соединением. На применении лазерных методов основаны разработки следующих процессов:
– AVLIS (Atomic Vapor Laser Isotope Separation) – США, Россия, Япония;
– SILEX (Separation of Isotopes by Laser Excitation ) – США совместно с Австралией;
– SILVA (Sèparation Isotopique par Laser sur Vapeur Atomique) – Франция;
– MLIS (Molecular Laser Isotope Separation) – США, Россия, Япония, ЮАР;
– CRISLA (Chemical Reaction Isotope Separation by Laser Activation) – США.
В США технология AVLIS была готова к промышленному освоению и к 2005 г. планировалось построить завод производительностью 1 млн. кг ЕРР/год. Однако в 1999 г. работы по программе AVLIS были прекращены из-за финансовых и технических трудностей. Вслед за США Франция отказалась от разработок процесса SILVA, который является аналогом AVLIS.
В табл. 1.3.11 приведено сравнение разных процессов обогащения урана. Анализируя состояние разработок новых методов и тенденций на рынке обогатительных услуг, можно сделать вывод, что в ближайшее время новые процессы вряд ли будут внедряться в промышленное производство. Базовыми промышленными технологиями обогащения урана останутся центрифугирование и газовая диффузия.
Мировые обогатительные мощности в настоящее время, как показано в табл. 1.3.12 составляют около 50 млн. кг ЕРР/год. Учитывая то обстоятельство, что спрос на обогатительные услуги в 2000 г. составлял ~ 35 млн. кг ЕРР/год, а в 2010 г. ожидается на уровне 37-43 млн. кг ЕРР/год, этих производственных мощностей вполне достаточно для обеспечения потребностей ядерной энергетики в ближайшей перспективе. 97 % всех мощностей по обогащению урана принадлежат четырем кампаниям: USEC, Eurodif, Urenco и Tenex.
Цена услуг на обогащение урана по долгосрочным контрактам в 2000 г. была около 80 долл./кг ЕРР. По разным прогнозам, в 2005 г. цены останутся либо на прежнем уровне, либо, в зависимости от обстоятельств, увеличатся до 85-90 долл./кг ЕРР.
3.3.2 Состояние и перспективы разделительных производств
в России
К настоящему времени разделение изотопов урана получило развитие в передовых развитых странах мира, таких, как США, Россия, Франция, Великобритания, Германия, Нидерланды, Япония, Китай.
Более 95 % мощностей мировой разделительной промышленности базируется на двух молекулярных методах: газовой диффузии и центробежном. Имеются полупромышленные установки (Бразилия, ЮАР), работа которых основана на эффекте разделения изотопов в сверхзвуковой струе газа. В США проведены крупномасштабные разработки по лазерному разделению изотопов урана на парах металлического урана, доведенные до создания полупромышленного модуля большой мощности. Однако возникшие технические трудности привели к прекращению работ по этому методу.
3.3.2.1 Развитие разделительной промышленности в СССР
и России
Разделительная промышленность СССР была создана в конце 40-х годов для производства оружейного урана с обогащением по 235U до концентрации 90 %. Первый промышленный завод был создан на основе газодиффузионного метода разделения. В течение короткого исторического периода (1945-1963 гг.) были введены в эксплуатацию четыре газодиффузионных завода, продукция которых шла на изготовление ядерного оружия, а также обеспечение ядерным топливом энергетических установок атомного флота. Часть обогащенного урана направлялась на мирные цели для снабжения ядерным топливом нарождающейся ядерной энергетики. В 60-х гг. велись интенсивные работы по реализации газоцентрифужного метода разделения изотопов урана, и в 1962 г. был пущен в эксплуатацию первый в мире газоцентрифужный завод. Преимущества этого метода, главным образом, его малая энергоемкость (примерно в 20 раз меньше, чем для метода газовой диффузии) позволили отказаться от газовой диффузии и постепенно переоснастить большинство диффузионных корпусов газовыми центрифугами.
Таблица 1.3.11
Сравнение процессов обогащения урана
| Процесс обогащения | Страна | Удельное потребление энергии, кВт·ч/кг ЕРР | Обогащение на одной ступени | Производительность | Технологическое давление | Количество материала, задействованного в процессе одновременно |
| Газовая | США, | 3000 (до 1978 г.) 2500 (после модернизации) | Низкое | Высокая | Умеренное | Большое (десятки тысяч тонн) |
| Франция | 2400 | |||||
| Газовые | СССР-Россия | 120-140 | Высокое | Низкая | Низкое | Малое |
| Великобритания, Нидерланды, Германия (фирма Urenco) | 100, новые модели 50 | |||||
| Газодинамические процессы: сопло вихревая трубка | Германия ЮАР | 3300 3500 | Среднее | Средняя | Умеренное | Малое |
| Химический обмен: Chemex-процесс | Франция | 600 | Среднее | Высокая | Низкое | Большое |
| Лазерный метод: AVLIS | США | 100-130 | Очень высокое | Очень низкая | Очень низкое | Малое |
Таблица 1.3.12
Мощности заводов по обогащению урана по состоянию на 2003 г.
| Страна | Местоположение завода | Организация, владеющая или управляющая заводом | Мощность, млн. кг ЕРР/год |
|
Газодиффузионная технология | |||
| США | Падука, шт. Кентукки | USEC | 11,3 |
| Портсмут, шт. Огайо (закрылся в 2001 г.) | 7,4 | ||
| Франция | Трикастен | Eurodif | 10,8 |
| Китай | Ланчжоу | CNNC | 0,9 |
| Всего (газовая диффузия) | 30,4 | ||
|
Центрифужная технология | |||
| Россия | Новоуральск | Минатом (Tenex) | 7,0 |
| Северск | 4,0 | ||
| Зеленогорск | 3,0 | ||
| Ангарск | 1,0 | ||
| Великобритания | Капенхерст | Urenco | 2,44 |
| Нидерланды | Алмело | 1,95 | |
| Германия | Гронау | 1,46 | |
| Япония | Рокасё | JNFL | 1,05 |
| Нингайо Тоге | JNC | 0,2 | |
| Китай | Ланчжоу | CNNC | 0,5 |
| Ханжонг | 0,5 | ||
| Пакистан | Кахута | PAEC | 0,005 |
| Всего (центрифугирование) | 23,1 | ||
| Суммарная мощность всех заводов | 53,5 | ||
В настоящее время Россия обладает самыми большими разделительными мощностями газоцентрифужных заводов в мире, что позволяет ей успешно конкурировать на международном рынке обогащенного урана, эффективно работать в изменившихся экономических условиях и использовать ранее накопленные богатые отвалы как альтернативу дефицита природного урана. За прошедшие годы были разработаны и пущены в серийное производство семь поколений газовых центрифуг, из которых последние три эксплуатируются по настоящее время.
3.3.2.2 Современное состояние и оборудование разделительных заводов
В общем объеме установленных на предприятиях России газовые центрифуги 5-го поколения составляют около 48 %, 6-го – 49 %, 7-го – 3%. В настоящее время на предприятиях проводится плановая замена отработавших ресурс газовых центрифуг 5-го поколения на центрифуги 7-го поколения. К 2010 г. газовые центрифуги 5-го поколения исчерпают ресурс, и в дальнейшем потребуется заменять выработавшие ресурс газовые центрифуги 6-го поколения.
Для производства центрифуг используются высокопрочные алюминиевые сплавы, высокоточные штамповки и трубные заготовки, упрочняющие волокна для роторных деталей (армос, стекловолокно), ужестчающие угольные композиции для ротора, искусственные лейкосапфиры для опорных подшипников, специальное литье из цветных металлов. Осуществляется широкая кооперация предприятий, изготавливающих исходные материалы и комплектующие изделия.
На предприятиях, изготавливающих газовые центрифуги, разработаны и внедрены специальные высокоточные станки для механической обработки роторных деталей, специальные намоточные программируемые станки, печи для полимеризации обмотки роторов, специальные сборочные конвейеры роторов и агрегатов, серия обкаточных и испытательных стендов.
На разделительных газоцентрифужных заводах используются высокоразвитая двухуровневая автоматизированная система управления технологическим процессом с контролем нескольких миллионов параметров как глобальных по всему заводу, так и по каждой центрифуге, процессом в аварийных ситуациях и при полном отключении внешнего электроснабжения. На заводах используются специальные датчики контроля параметров процесса.
Для электроснабжения двигателей газовых центрифуг применяются специализированные статические преобразователи, вырабатывающие электропитание повышенной частоты с высокой степенью ее стабилизации, не зависящие от колебаний напряжения и частоты во внешней электросети. В корпусах газоцентрифужных заводов, суммарный объем которых превышает несколько миллионов м3, постоянно поддерживается температура 18-20 °С. Современные газоцентрифужные заводы представляют собой полностью автоматизированные предприятия, работающие круглосуточно в течение ресурсного срока, достигающего для последних поколений газовых центрифуг 25-30 лет.
Технологический процесс разделения осуществляется при низком давлении (5-40 мм рт. ст., абс.), что обеспечивает отсутствие каких-либо выделений радиоактивных и химически вредных веществ в рабочие помещения, поддержание выбросов в окружающую среду на уровне ниже санитарных норм.
Дата: 2019-02-25, просмотров: 517.