Производство гексафторида урана из высших оксидов урана
И стадию получения тетрафторида урана
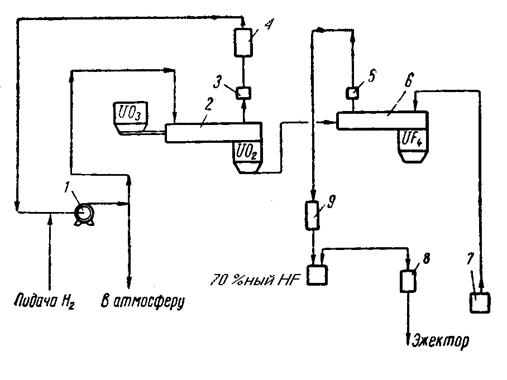
Принципиальная технологическая схема производства гексафторида урана, применяемая на зарубежных заводах, приведена на рис. 1.3.1.

Рис. 1.3.1 Технологическая схема производства гексафторида урана
на зарубежных предприятиях
Процесс восстановления высших оксидов водородом описан в главе 2.
Тетрафторид урана служит важнейшим промежуточным продуктом, используемым для производства гексафторида урана и металлического урана.
3.1.1 Термодинамика и кинетика гидрофторирования диоксида
урана
Реакция гидрофторирования диоксида урана безводным фтороводородом описывается уравнением:
UO2(тв) + 4HF = UF4(тв) + 2H2O + Q, Q = 266,635 кДж/моль. (1.3.5)
Как видно из уравнения, процесс является обратимым; для него характерен значительный тепловой эффект. Более подробно термодинамические особенности процесса фторирования UO2 безводным фтороводородом описаны в специальной научно-технической литературе. Термодинамику этого процесса можно также рассчитывать с помощью компьютерных программ ASTRA, CVD и “Химический верстак”, более подробные сведения о которых приведены в главе 2 части I учебного пособия.
По закону действия масс константа равновесия реакции гидрофторирования выражается уравнениями:
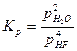 ;
; 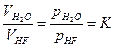 . (1.3.6)
. (1.3.6)
Если 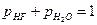 атм, то
атм, то 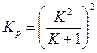 . (1.3.7)
. (1.3.7)
Равновесное содержание фтороводорода в газовой фазе над смесью тетрафторида и диоксида урана характеризуется данными, приведенными в табл. 1.3.1.
Таблица 1.3.1
Температурная зависимость равновесного давления паров воды
и фтороводорода над тетрафторидом урана
| Температура, °С | Равновесное давление паров, атм | Соотношение
| Константа гидролиза, К | |
| HF | Н2O | |||
| 200 | 0,014 | 0,986 | 1,42·10–2 | 4,0·10–8 |
| 250 | 0,020 | 0,980 | 2,04·10–2 | 1,4·10–7 |
| 300 | 0,036 | 0,964 | 3,73·10–2 | 1,4·10–8 |
| 400 | 0,085 | 0,915 | 9,29·10–2 | 6,2·10–6 |
| 500 | 0,160 | 0,840 | 19,00·10–3 | 1,0·10–3 |
| 600 | 0,574 | 0,426 | 135,00·10–2 | 5,9·10–1 |
| 700 | 0,823 | 0,177 | 460,00·10–2 | 1,510–1 |
При анализе термодинамических данных можно сделать вывод о том, что тетрафторид урана легко разлагается водяным паром. Следовательно, для увеличения полноты реакции гидрофторирования диоксида урана необходим избыток фтороводорода.
Так как реакция гидрофторирования диоксида урана обратима, причем вероятность протекания обратной реакции увеличивается с нагреванием, повышение температуры процесса целесообразно только в случае, если в реакционной смеси газов концентрация фтороводорода высока по отношению к содержанию паров воды. В присутствии большого количества водяных паров (на выходе из реактора в противоточном процессе) необходимо поддерживать сравнительно низкую температуру. Это условие можно выполнить, если процесс гидрофторирования разделить на несколько стадий. В соответствии с данными о равновесной концентрации фтороводорода в смеси с парами воды, гидрофторирование диоксида урана выгодно проводить при возможно более низких температурах. Однако при этом существенно снижается скорость процесса. На практике процесс гидрофторирования проводят при 400-600 °С.
Скорость взаимодействия диоксида урана с фтороводородом определяется рядом факторов. Наибольшее влияние оказывает температура. При ее увеличении скорость реакции гидрофторирования возрастает. Влияние на скорость процесса также оказывает концентрация фтороводорода. Разбавление фторирующего реагента инертным газом приводит к снижению скорости процесса. Еще больше снижается скорость реакции при введении в систему паров воды.
Так как взаимодействие диоксида урана с фтороводородом – гетерогенная реакция, кинетические зависимости в этом случае определяются также физическими и физико-химическими характеристиками диоксида и тетрафторида урана. Установлено, что различные образцы диоксида урана обладают разной активностью по отношению к реакции гидрофторирования. Одни из них гидрофторируются очень быстро даже при сравнительно низкой температуре, но процесс, как правило, не доходит до конца. Другие, наоборот, характеризуются более низкой скоростью процесса, но при этом наблюдается количественное превращение диоксида в тетрафторид.
Для правильного понимания этих различий необходимо учитывать разницу в размерах кристаллов и агрегатов диоксида урана в удельной поверхности порошков и т.д. Кроме того, вследствие значительного выделения тепла при гидрофторировании и плохой теплопроводности смеси UO2-UF4 возможно оплавление и спекание наружной поверхности частиц, что затрудняет доступ газообразного фтороводорода к внутренним слоям материала. Поэтому дальнейшее гидрофторирование зависит от скорости диффузии фтороводорода через наружный слой тетрафторида урана. Процесс спекания может быть обусловлен образованием эвтектик между диоксидом и тетрафторидом урана. Эвтектические составы с 12 и 18 % мол. диоксида имеют точки плавления соответственно 886 и 930 °С. Температура спекания зависит от гранулометрического состава материала и может быть намного ниже температуры плавления, составляя в ряде случаев 500-600 °С.
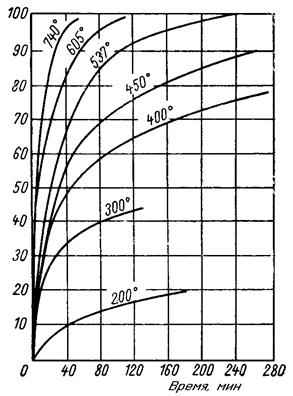
Количественное рассмотрение кинетических закономерностей реакции гидрофторирования начнем с классификации (конечно, несколько условной) образцов диоксида урана по удельной поверхности и размерам агрегатов. Из всего многообразия порошков можно выделить три типа: тонкий продукт, крупный пористый продукт, представляющий собой конгломерат тонких частиц и крупный непористый продукт. При гидрофторировании крупных непористых частиц образующийся на их поверхности тетрафторид защищает материал от дальнейшего воздействия HF; повышение температуры не приводит к разрушению этого защитного слоя. При гидрофторировании крупного пористого материала повышение температуры, связанное с увеличением эффекта диффузии, приводит к увеличению степени реагирования; возникающее при этом спекание кристаллов тетрафторида урана обусловливает максимум на кривых. При гидрофторировании тонкого диоксида эффект спекания частиц еще не наблюдается (для определенного размера частиц и температуры), и он реагирует практически нацело. Отмеченные закономерности характеризуются данными, приведенными на рис. 1.3.2.
|
Рис. 1.3.2 Поведение различных порошков UO2 при гидрофторировании газообразным фтороводородом; длительность процесса 1,5 ч. Образцы получены по реакции UO2(NO3)2 |
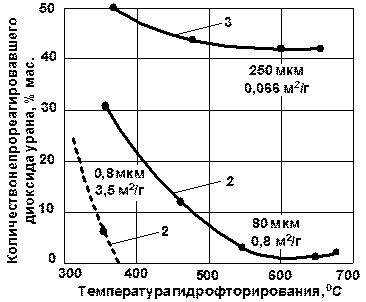
 UO3
UO3  UO2
UO2В том случае, если процесс гидрофторирования не осложнен эффектами, связанными со спеканием и оплавлением материала, повышение температуры приводит к значительному увеличению скорости процесса (рис. 1.3.3). Энергия активации (Еа) этого процесса колеблется в зависимости от многих факторов в довольно широких пределах, составляя в большинстве случаев 25,1-62,9 кДж/моль. Повышение концентрации фтороводорода приводит к заметному увеличению скорости гидрофторирования лишь при сравнительно низкой температуре, около 400 °С; при более высокой температуре (600 ° С и выше при большей концентрации фтороводорода) эффект этот в значительной мере смазывается за счет наблюдающегося в этих условиях спекания материала.
Разбавление фтороводорода парами воды также приводит к уменьшению скорости гидрофторирования. Но одновременно отмечается для каждого данного состава газовой фазы оптимальная температура, при которой достигается одинаковая степень превращения диоксида yранa в тетрафторид за минимальное врeмя (рис. 1.3.4). Как видно из этих данных, повышение температуры гидрофторирования для всех составов газовой фазы вначале приводит к уменьшению времени, необходимого для 75 % реагирования; однако при дальнейшем возрастании температуры (выше максимума, характерного для каждого заданного состава газовой фазы) необходимое время процесса гидрофторирования не уменьшается, а увеличивается, что объясняется увеличивающейся в этом случае скоростью реакции гидролиза тетрафторида урана.
|
Парциальное давление фтороводорода 1 aтм; пары воды отсутствуют. Диоксид урана получен восстановлением кристаллического триоксида урана в водороде при 750 °С. Рис. 1.3.3 Влияние температуры на скорость гидрофторирования диоксида урана |
| Прореагировало UO2, % мас. |
С увеличением температуры гидрофторирования происходит рост частиц тетрафторида урана. Резкое увеличение размеров частиц при фто-рировании наблюдается для самых тонких образцов диоксида; при температуре выше 600 °С размер кристаллов тетрафторида уже мало зависит от дисперсного состава исходного продукта. Спекание частиц, связанное с увеличением температуры, приводит к возрастанию насыпной массы тетрафторида. Значительнoe увеличение насыпной массы наблюдается для тонких фракций диоксида урана.
При сравнительно низкой температуре (450 °С), когда эффект спекания еще не имеет значения, степень реагирования диоксида урана всецело обусловлена поверхностью соприкосновения твердой и газовой фаз и максимальна для образцов диоксида, имеющего самые тонкие частицы. При более высокой температуре (650 °С) диоксид урана, состоящий из тонких частиц, склонных к спеканию, фторируется хуже, чем диоксид, состоящий из более крупных частиц.
|
Диоксид урана получен восстановлением триоксида урана водородом Рис. 1.3.4 Влияние разбавления фтороводорода парами воды на время, в течение которого достигается 75% реагирования |
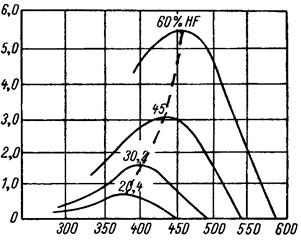
| Величина, обратная времени достижения 75 %-ного выхода UF 4 , ч–1 |
| Температура, °С |
Эффективность гидрофторирования диоксида урана определяется реакционной способностью исходного продукта, зависящей в основном от температуры его получения. Диоксид урана, полученный при низких температурах, характеризуется высокой реакционной способностью, что подтверждается его пирофорностью (способностью самовозгораться на воздухе). На рис. 1.3.5 приведены кривые, характеризующие степень гидрофторирования диоксида урана во времени в зависимости от температуры его получения. Диоксид урана был получен восстановлением триоксида водородом. Триоксид урана получали из уранилнитрата. Во всех трех случаях диоксид урана обрабатывали газорбразным фтороводородом при 575 °С. Использование диоксида урана, полученного при низкой температуре, позволяет значительно снизить избыток фтороводорода при гидрофторировании.
Химическая активность диоксида урана определяется, кроме того, его физическими характеристиками: удельной поверхностью, размером кристаллов и агрегатов и т.д. На практике, однако, количественное соотношение между скоростью гидрофторирования и этими свойствами диоксида урана не установлено. Условная классификация порошков диоксида урана, приведенная ранее, конкретизируется по методам получения исходного продукта. Порошки первого типа (крупный непористый продукт), полученные денитрацией уранилнитрата до триоксида и последующим восстановлением водородом или аммиаком до диоксида, обладают относительно малой активностью. Для гидрофторирования такого продукта требуются повышенные температуры, в конечных продуктах обычно находится некоторое количество непрореагировавшего диоксида. Порошки диоксида урана второго типа (крупный пористый продукт), полученные аналогично порошкам первого типа, но в условиях непрерывного перемешивания при денитрации, более активны, но при их гидрофторировании также необходимы высокие температуры.
| Количество прогидрофторированного UO 2 , % мас. |
Время, мин
|
Температура гидрофторирования – 575 °С. Рис. 1.3.5 Зависимость скорости процесса гидрофторирования диоксида урана газообразным фтороводородом от температуры его получения из триоксида |
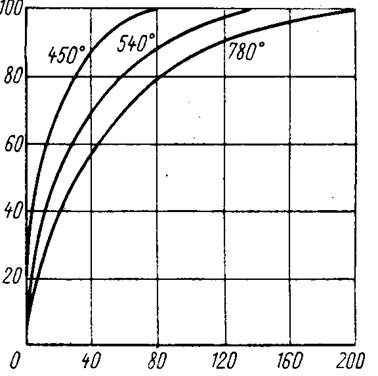
Порошки диоксида урана третьего типа, полученные из триоксида, с величиной зерна 1 мкм наиболее полно гидрофторируются при умеренных температурах. Диоксид урана, полученный из полиураната аммония, благодаря своей мелкозернистости гидрофторируется достаточно быстро при сравнительно низких температурах. Однако этот продукт склонен к спеканию, в результате чего некоторая часть его остается непрофтори-рованной. Диоксид урана, полученный из полиуранатов аммония с предварительным их превращением в закись-окись и ее восстановлением водородом или аммиаком, состоит из агломератов частиц, размер которых больше, чем у частиц диоксида, непосредственно полученного из полиуранатов аммония. Активность этого диоксида урана несколько ниже.
Скорость гидрофторирования зависит также от химического состава диоксида. Так, при увеличении содержания шестивалентного урана в продукте до 3 % скорость реакции удваивается. Удельная поверхность тетрафторида урана, полученного из частично окисленного диоксида, примерно вдвое больше, чем у тетрафторида, полученного из того же диоксида урана без окисления, хотя заметной разницы в удельной поверхности исходных продуктов нет. Причины увеличения скорости реакции гидрофторирования при частичном окислении диоксида урана не изучены, но известно, что в процессе обработки такого продукта газообразным фтороводородом оплавления или спекания материала не наблюдается.
Скорость гидрофторирования диоксида урана повышается также от содержания таких примесей, как SО  ион, сульфат аммония и понижается от присадки натрия.
ион, сульфат аммония и понижается от присадки натрия.
Рассмотренные закономерности относятся к процессу гидрофторирования диоксида урана в неподвижном слое, в постоянном токе фтороводорода, а следовательно, в большом его избытке. На практике процесс целесообразно проводить так, чтобы расход фтороводорода был минимальным. Для этого во всех применяемых в настоящее время конструкциях аппаратов осуществляется противоток твердой и газообразной фаз. Свежий диоксид урана на входе в аппарат гидрофторируется газом, содержащим значительное количество паров воды; напротив, в существенной степени профторированный продукт на выходе из аппарата обрабатывают фтороводородом, не содержащим паров воды. Таким образом по длине реакционного аппарата устанавливается определенный градиент концентрации тетрафторида урана и диоксида урана в твердой фазе, а также фтороводорода и паров воды в газовой фазе.
Использование принципа прямотока твердой и газообразной фаз в любом случае вызовет повышенный расход фтороводорода. С этим можно было бы не считаться, если бы отделение фтороводорода от воды не было бы затруднено. Тогда часть фтороводорода циркулировала бы в процессе, а расход реагента был бы близок к стехиометрически необходимому. Однако отделить фтороводород от воды сложно, и при простой конденсации газов после гидрофторирования фтороводород в значительной степени обесценен наличием воды.
Из термодинамических соотношений следует, что, чем выше температура, тем большее количество тетрафторида урана гидролизуется выделяющимися парами воды и тем, следовательно, большая концентрация фтороводорода необходима для подавления гидролиза. Таким образом, для максимального использования фтороводорода выгодно проводить процесс при возможно низкой температуре. Однако в этих условиях скорость реакции невелика и процесс будет протекать очень длительно. Доказано, что при постепенном повышении температуры в ходе гидрофторирования достигается более высокая общая скорость реакции, чем при поддержании только лишь высокой температуры, которая вызывает частичное спекание твердого продукта и уменьшение скорости реакции.
Указанные обстоятельства обусловили необходимость создания в аппаратах гидрофторирования градиента температуры; на выходе твердого продукта из реактора температура максимальна, на входе в реакционный аппарат она минимальна. В свежем фтороводороде, поступающем на гидрофторирование, пары воды отсутствуют и температура в этой части реактора может быть весьма значительной (500-600 °С), что обусловливает высокую скорость процесса. По мере увеличения содержания паров воды в газовой фазе температуру реакционного пространства необходимо снизить, и на входе твердого вещества она может составлять 300-400 °С; здесь аппарат работает лишь для улавливания и связывания непрореагировавшего ранее фтороводорода.
В идеальном случае градиент температуры в реакционном пространстве должен быть постоянным. На практике процесс осуществляют, как, правило, в нескольких аппаратах, в которых поддерживают различную температуру.
Необходимость поддержания определенного градиента температуры по ходу гидрофторирования, а также осуществление принципа противотока твердого вещества и газа обусловлено также высоким тепловым эффектом реакции. Если свежий диоксид урана обрабатывать безводным фтороводородом (при прямотоке) и процесс этот сразу осуществлять при достаточно высокой температуре (отсутствует градиент температуры), выделяющееся в ходе процесса тепло невозможно отвести от системы, что приводит к повышению температуры твердого материала и, как следствие, к его спеканию и оплавлению. Напротив, использование противотока твердого вещества и газа, создание температурного перепада в реакционном пространстве позволяет управлять реакцией и добиться высокой эффективности гидрофторирования.
В диоксиде урана, используемом для гидрофторирования, всегда в большем или меньшем количестве присутствуют высшие оксиды урана: триоксид или закись-окись. Взаимодействие триоксида урана с фтористым водородом протекает при сравнительно низкой температуре (100-200 °С) по реакции:
UO3 + 2HF → UO2F2 + H2O, (1.3.8)
причем выделяющиеся в этом процессе пары воды сравнительно мало сдвигают равновесие в обратную сторону. Гидрофторирование триоксида урана – процесс значительно менее обратимый, чем гидрофторирование диоксида.
Закись-окись урана при обработке фтороводородом переходит в смесь тетрафторида урана и уранилфторида:
U3O8 + 8HF → UF4 + 2UO2F2 + 4H2О. (1.3.9)
Оксиды урана с составом, промежуточным между диоксидом и закисью-окисью, ведут себя аналогично, т.е. при гидрофторировании образуют смесь тетрафторида и уранилфторида в строгом соответствии с соотношением четырех- и шестивалентного урана в исходном продукте. Скорость гидрофторирования зависит от условий приготовления образцов; с повышением температуры изготовления их активность, как правило, уменьшается.
Таким образом, наличие шестивалентного урана в диоксиде (как в форме триоксида, так и в форме закиси-окиси) будет приводить к образованию уранилфторида. В связи с высокой скоростью гидрофторирования высших оксидов урана (особенно при низкой температуре) практически можно считать, что весь шестивалентный уран в тетрафториде присутствует только в форме уранилфторида.
3.1.2 Режим и аппаратурное оформление гидрофторирования
диоксида урана фтороводородом
В первые годы эксплуатации урановых заводов производство тетрафторида методом гидрофторирования осуществлялось периодически. Триоксид урана в лотках из нержавеющей стали помещали в печь и подвергали восстановлению. Полученный диоксид урана охлаждали в атмосфере азота и перегружали вручную в монелевые, графитовые или магниевые лотки; затем диоксид урана обрабатывали газообразным фтороводородом при 500 °С в течение 20-30 ч. Реактор, изготовленный из высококачественной жаропрочной стали, имел форму бокса. Лотки с продуктом устанавливали и вынимали из реактора через дверцу, охлаждаемую водяной рубашкой. Реакторы объединяли обычно в серии (по 5-15 штук) так, чтобы газы проходили последовательно из первого реактора во второй и т.д.
В каждый лоток загружали 30-35 кг диоксида урана. Получаемый продукт, содержал 97 % тетрафторида урана. При таком процессе продукт получался хорошего качества, но стоимость его была высокой. Кроме того, для выполнения операций загрузки и разгрузки в условиях периодического процесса требовались защитное оборудование и защитная одежда для предохранения людей от воздействия урановой пыли.
Возросшая потребность в уране вызвала необходимость замены периодических методов получения тетрафторида урана более эффективными, высокопроизводительными непрерывными процессами. Наиболее значительным усовершенствованием способа гидрофторирования диоксида урана оказалось создание горизонтальных шнековых реакторов с вибрирующим лотком, а также реакторов для гидрофторирования в псевдоожиженном слое и вертикальных реакторов для гидрофторирования таблетированного или гранулированного диоксида урана.
3.1.2.1 Гидрофторирование в реакторах со шнековым
перемешиванием
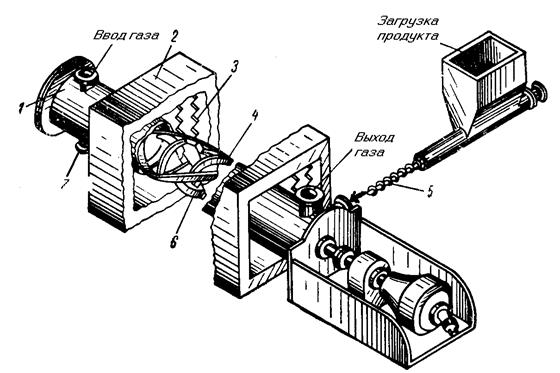
Общая схема процесса производства тетрафторида урана из триоксида представлена на рис. 1.3.6. Диоксид урана, полученный при восстановлении триоксида водородом, обрабатывают безводным фтороводородом; процесс осущестляют по принципу противотока. На рис. 1.3.7 показано устройство реактора со шнековым перемешиванием для гидрофторирования диоксида урана. Горизонтальная труба реактора выполнена из инконеля или из сплава хастеллой. Внутри трубы имеется мешалка, конструкция которой определяется условиями гидрофторирования. При обработке диоксида урана с низкой реакционной способностью для полного перевода его в тетрафторид требуется высокая температура; в этом случае мешалка предназначена главным образом для предотвращения спекания твердого продукта и выполняется без центрального вала с лентами, изогнутыми в спираль.
|
1-газодувка для Н2; 2-реактор для восстановления; 3,5-фильтры; 4,9-конденсаторы; 6-реактор для гидрофторирования; 7-испаритель HF; 8-скруббер Рис. 1.3.6 Схема получения тетрафторида урана из триоксида
|
Если диоксид урана обладает высокой реакционной способностью, гидрофторирование осуществляют при более низкой температуре в течение меньшего промежутка времени; в этом случае мешалка главным образом транспортирует материал. Скорость вращения мешалки колеблется в пределах 4-20 об/мин; она перемещает твердый продукт внутри реакционной трубы, перемешивает и разгребает его, улучшая таким образом контакт между твердой и газовой фазами. В большинстве случаев оптимальным является следующий температурный режим: равномерный подъем температуры по длине реактора (от загрузочного конца) от 300 до 550 °С.
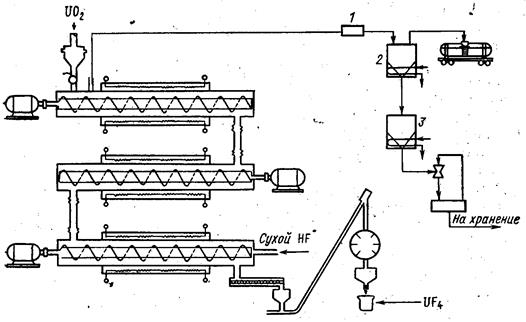
На заводах в Падьюке и Уэлдон-Спринге (США) тетрафторид урана получают непрерывно в трех последовательно соединенных шнековых реакторах, расположенных горизонтально один над другим. В этом случае коффициент использования фтороводорода превышает 90 %. Технологическая схема такого процесса приведена на рис. 1.3.8. Труба каждого реактора изготовлена из инконеля диаметром 400 мм и длиной более 6 м. Для удобства обслуживания трубы снабжены на концах фланцами, обогреваются электрическими печами, имеющими зоны нагрева и охлаждения. Охлаждение осуществляют воздухом.
|
1-корпус реактора; 2-печь; 3-элементы печи; 4-распорки; 5- шнек для питания; 6-лопасти; 7-разгрузочное отверстие Рис. 1.3.7 Схема реактора гидрофторирования двуокиси урана со шнековым перемешиванием |
Верхний реактор загружают диоксидом либо непосредственно из реактора восстановления, либо из промежуточного бункера. В обоих случаях продукт подают в небольшой запорный бункер, из которого винтовым конвейером он направляется в реактор гидрофторирования. Порошок диоксида, так же как в одноступенчатом аппарате, перемещается при помощи шнека-мешалки. Скорость подачи диоксида урана зависит от его активности. Для обычного промышленного диоксида урана, полученного из уранилнитрата через триоксид, скорость вращения мешалки в первых двух реакторах равна 8 об/мин, в последнем реакторе 12 об/мин. Производительность установки в этом случае составляет около 200 кг/ч тетрафтодида урана.
Качество получаемого тетрафторида урана определяется реакционной способностью диоксида урана, продолжительностью гидрофторирования и изменением температуры в ходе процесса. Типичный анализ продукта (в %), получаемого в трехступенчатом реакторе со шнековым перемешиванием, следующий:
| Общий U | 76,0 | Ni | 0,0035 |
| UF4 | 96,2 | Cr | 0,0009 |
| UO2F2 | 2,0 | Mn | < 0,001 |
| Оксиды урана | 1,8 | Cd | < 0,00005 |
| Fe | 0,0055 | Mo | < 0,001 |
|
1-фильтр; 2-конденсатор для 70 %-ного HF; 3- конденсатор для безводного HF Рис. 1.3.8 Схема установки для гидрофторирования диоксида урана в шнековых реакторах
|
Насыпная масса продукта с утряской равна 3,5 г/см3. Температура в процессе гидрофторирования постепенно повышается с 300 °С в месте загрузки диоксида урана до 550 °С на выгрузке тетрафторида; спекания твердого порошкообразного продукта не происходит; продолжительность процесса обычно составляет 4-5 ч.
Безводный фтороводород (99,95 % HF), используемый в этом процессе, подаются за счет давления собственных паров или при помощи сжатого воздуха в испаритель с паровой рубашкой. Перегретые пары фтороводорода поступают с определенной скоростью в реактор для гидрофторирования. Отходящие газы проходят через циклон, где отделяется твердая фаза, затем их дважды фильтруют через монелевые и угольные фильтры, и направляют в конденсатор. Пыль, осевшая на фильтрах, через определенные промежутки времени выдувают обратно струёй сжатого азота. В трубчатом конденсаторе при 65 °С конденсируется 70 %-ная плавиковая кислота, стекающая в сборник. Безводный фтороводород, поступающий из трубчатого конденсатора, охлаждается до – 40 °С с целью его количественного улавливания. Оставшиеся газы выбрасываются в атмосферу воздушным эжектором.
Необходимо отметить, что при конденсации фтороводорода в каком-либо месте реактора или фильтрующей системы возникает серьезная опасность коррозионного разрушения. Поэтому места, где возможна конденсация, необходимо обогревать при помощи паровых рубашек или электронагревателей. Тетрафторид урана перед взвешиванием охлаждают азотом в шнековом транспортере до 150 °С и при помощи конвейера пересыпают в бункер, являющийся промежуточной емкостью перед подачей его на получение металлического урана или гексафторида.
Процесс гидрофторирования диоксида урана можно осуществлять во вращающейся печи при 450 °С в потоке предварительно нагретого до 250 °С фтороводорода; избыток фтороводорода составляет 100 %. Отходящие газы нейтрализуются в системе скрубберов, по которым циркулирует 5 %-ный раствор гидроксида натрия или калия. К газообразному фтороводороду добавляют некоторое количество водорода, который, являясь восстановителем, препятствует образованию в получаемом продукте уранилфторида.
Осуществление непрерывного процесса гидрофторирования во вращающейся печи связано с трудностями, заключающимися в уплотнении вращающихся частей печи. Эти трудности преодолимы применением сальников, работающих под избыточным давлением углекислого газа, поддерживаемым между внутренним уплотнительным кольцом из тефлона и внешним кольцом из резины.
Получаемый тетрафторид урана имеет насыпную массу, равную 1,9 г/см3; содержание диоксида урана и уранилфторида в конечном продукте не превышает 0,5 %.
Подобные печи рекомендованы для производства тетрафторида урана в больших масштабах.
Обогащение урана
3.3.1 Способы разделения изотопов урана и мощности заводов
по разделению изотопов урана в мире
Природный уран содержит около 0,7 % делящегося изотопа 235U, тогда как для работы современных LWR требуется концентрация этого изотопа 3-5 %.
Процессы обогащения урана по 235U основаны на разности масс атомов 235U и 238U и связанных с этим небольших различиях в физико-химических свойствах. На основе этих различий можно проводить разделение изотопов с помощью газовой диффузии, центрифугирования, газодинамических процессов, химического обмена, лазерными методами и т.д.
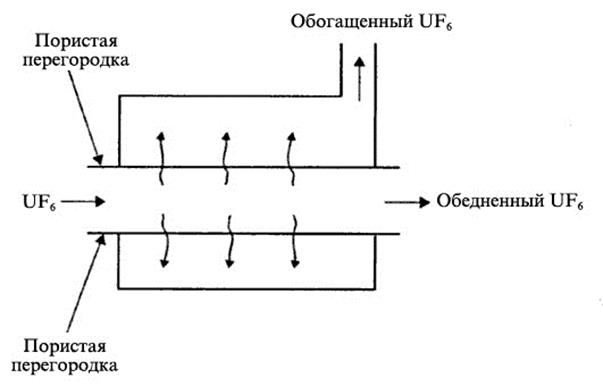
Исторически первым процессом, широко использовавшимся для промышленного обогащения урана, был процесс газовой диффузии через пористые перегородки. Разделение изотопов с помощью газодиффузионного метода основано на различии в скоростях движения молекул газообразных соединений 235U и 238U. Как известно, в смеси газов все молекулы имеют одинаковую кинетическую энергию, а для этого, в соответствии с уравнением Е=1/2∙m∙V2, молекулы с более легкой массой должны двигаться быстрее и ударяться о стенки перегородки чаще, чем тяжелые молекулы. Если перегородка будет пористой, то легкие молекулы будут проходить через нее интенсивнее, и смесь за перегородкой будет обогащена ими.
Газообразное соединение урана, пригодное для проведения процесса обогащения, должно быть летучим и химически устойчивым. Химический элемент, входящий в состав данного соединения урана, должен иметь только один изотоп и обладать низкой атомной массой. Этим требованиям удовлетворяет гексафторид урана. При комнатной температуре и атмосферном давлении он является бесцветным твердым веществом. Температура сублимации UF6 составляет 56,4 ºС.
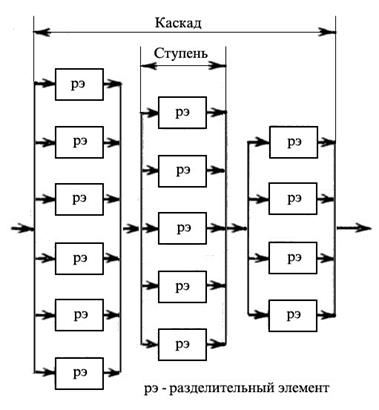
Схема диффузионного процесса через пористую перегородку показана на рис. 1.3.35. Массовые числа молекул 238UF6 и 235UF6 составляют 352 и 349, соответственно. Максимальный элементарный коэффициент разделения равен квадратному корню из отношения массовых чисел и составляет 1,0043. Однако на практике коэффициент разделения несколько меньше из-за различных технологических недостатков (в среднем около 1,001). Для достижения требуемой степени обогащения необходимо повторять процесс много раз, для чего разделительные элементы объединяются в ступени и каскады, показанные на рис. 1.3.36 и 1.3.37.
|
Рис. 1.3.35 Схема процесса газовой диффузии |
|
Рис. 1.3.36 Схема размещения разделительных элементов в ступенях и ступеней в сужающемся каскаде
|
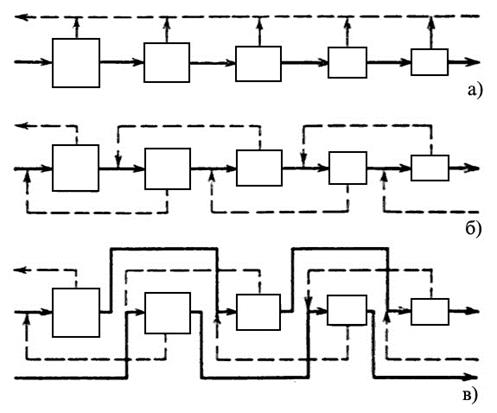
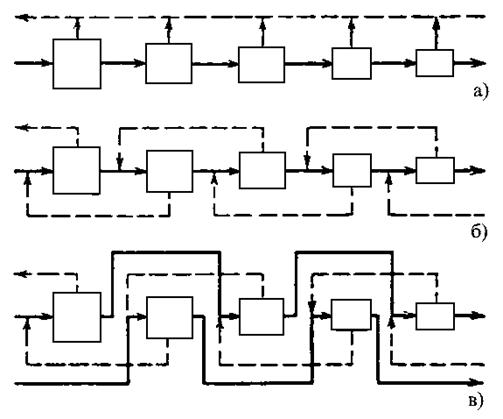
|
а) – простой каскад, б) – противоточный симметричный каскад, в) – противоточный несимметричный каскад с подачей потока питания через одну ступень в прямом направлении |
 – обедненные потоки;
– обедненные потоки; – обогащенные потоки;
– обогащенные потоки;Рис. 1.3.37 Примеры каскадов
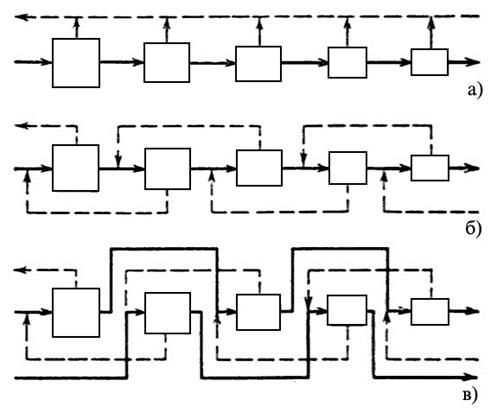
В простом каскаде ступени соединяются последовательно, и обогащенная фракция любой ступени служит питанием для следующей ступени. При этом обедненные фракции на каждой ступени считаются отвалом и дальнейшей переработке не подлежат. В противоточном каскаде обедненная фракция, выходящая в каждой ступени, направляется на переработку в предыдущую ступень. На обогатительных заводах обычно используются противоточные каскады, так как они позволяют добиться более высокого выхода продукта.
Стандартные элементы каскада на газодиффузионных заводах включают:
1. делители, содержащие диффузионные фильтры;
2. компрессоры, устанавливаемые на каждой ступени для создания давления, необходимого для прохождения газа через фильтры;
3. теплообменники для отвода теплоты сжатия газа;
4. электромоторы, приводящие в действие компрессоры и теплообменники;
5. трубопроводы внутренних и внешних коммуникаций, клапаны и т.п.
Диффузионные фильтры, находящиеся в делителе, должны иметь малую толщину и высокую пористость, при этом поры должны быть очень мелкими. Учитывая тот факт, что фильтры должны работать продолжительное время в коррозионноактивных условиях, материалы фильтров должны быть химически инертными и обладать высокой механической прочностью. В качестве материалов для изготовления фильтров могут использоваться металлы (Au, Ag, Ni, Al, Cu), оксиды, фториды и нитриды металлов, фторопласты. Изготовление фильтров осуществляется путем протравливания тонких пластин из выбранного материала или прессования тонких порошков материала с последующим спеканием.
|
Рис. 1.3.38 Сборка трубчатых газодиффузионных фильтров |

На рис. 1.3.38 и 1.3.39 показаны сборка с фильтрами и газодиффузионная ступень в каскаде. Типичный газодиффузионный каскад, производящий низкообогащенный уран для АЭС (обогащение не выше 5 %), включает более 1000 ступеней. Для работы газодиффузионного завода требуется большое количество электроэнергии, и поэтому стоимость продукта в значительной мере зависит от ее стоимости.
|
Рис. 1.3.39 Газодиффузионная ступень в каскаде |

Газодиффузионная технология обогащения использовалась на заводах СССР, США, Франции, Великобритании, Китая и Аргентины. До 1960-1970-х гг. она была основной технологией обогащения урана в мире. Затем газовая диффузия постепенно стала вытесняться более конкурентоспособной центрифужной технологией. В настоящее время газодиффузионная технология используется только в США, Франции и Китае. В США до недавнего времени работало два газодиффузионных завода: в Портсмуте, штат Огайо, и в Падуке, штат Кентукки. Срок их службы к концу ХХ века приближался к 50 годам, и нужно было решать их дальнейшую судьбу. В результате газодиффузионный завод в Портсмуте было решено закрыть, а в здании, где в 1980-е годы испытывали центрифуги, разместить центрифужный завод. Завод в Падуке будет модернизирован и продолжит свою работу.
Во Франции действует один газодиффузионный завод, расположенный в Трикастене. Завод является рентабельным благодаря использованию дешевой электроэнергии с АЭС Трикастен и будет работать до 2010 г. Однако и на этом заводе в будущем планируется заменить газодиффузионную технологию на центрифужную. Разработка центрифуг будет осуществляться компанией Eurodif совместно с компанией Urenco.
Обогащение урана с помощью метода центрифугирования основано на эффекте разделения изотопов сильным центробежным полем во вращающемся цилиндрическом роторе. При высоких угловых скоростях в условиях вакуума более тяжелые молекулы 238UF6 двигаются к периферии, обеспечивая частичное разделение изотопов в радиальном направлении. Если создать осевой (противоточный) поток во вращательном движении газа, то степень обогащения повысится за счет добавочного обогащения в осевом направлении. Создание противотока может быть осуществлено с помощью неподвижных экранов внутри ротора, температурных градиентов и внешних насосов. В результате происходящих в центрифуге процессов тяжелая фракция с 238U опускается в нижнюю часть, а легкая с 235U поднимается вверх.
Элементарный коэффициент разделения изотопов в газовой центрифуге гораздо выше, чем в газодиффузионной ячейке. Если в газодиффузионной ячейке он, как указано выше, составляет реально около 1,001, то в газовой центрифуге – 1,09. Это позволяет снизить число ступеней разделения на центрифужном заводе примерно в 100 раз по сравнению с газодиффузионным предприятием.
|
Рис. 1.3.40 Противоточная газовая центрифуга |
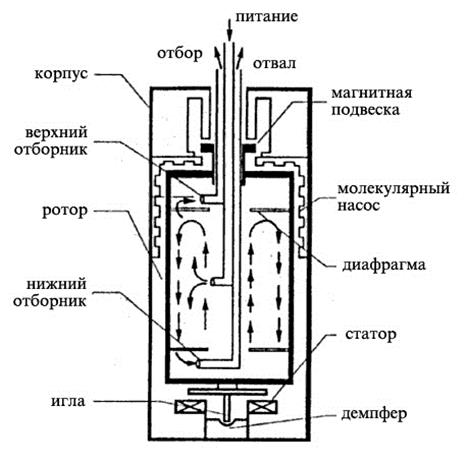
На рис. 1.3.40 изображена схема противоточной газовой центрифуги. В центре ротора установлены три концентрические трубы для подачи питания и отвода обогащенного продукта и отвала. Верхняя часть ротора удерживается в вертикальном положении с помощью магнитной подвески. Отбор продукта и отвала осуществляется вверху и внизу ротора, соответственно, с помощью газозаборников типа трубок Пито.
Производительность центрифуги в разделении изотопов пропорциональна длине ротора и скорости вращения в четвертой степени. По этой причине более предпочтительными являются центрифуги с возможно большей длиной ротора, работающие при высоких угловых скоростях. Длина ротора центрифуги ограничивается рядом практических соображений. Очень длинные роторы имеют тенденцию к изгибу, ведущему к неоднородностям газовых потоков. Кроме того, длинные роторы увеличивают стоимость оборудования для их изготовления, транспортирования роторов с заводов-изготовителей, а также стоимость производственных зданий.
Если газ с молекулярной массой М и молекулярной плотностью ρ подвергают центробежному ускорению ω2·r, то на единицу его объема действует сила Мρω2r. Здесь ω – угловая скорость и r – радиус. Градиент давления, установившийся в газе, составит:
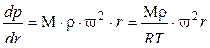 . (1.3.48)
. (1.3.48)
Для того чтобы найти отношение давлений, соответствующих радиусам 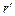 и
и 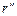 , уравнение (1.3.48) необходимо проинтегрировать. В результате получим:
, уравнение (1.3.48) необходимо проинтегрировать. В результате получим:
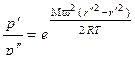 . (1.3.49)
. (1.3.49)
Полученная зависимость аналогична экспоненциальному закону изменения барометрического давления при изменении высоты.
Для смеси газов аналогичная зависимость имеет место для парциальных давлений каждого компонента. В бинарной смеси при равновесии:
– для легкого компонента
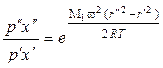 , (1.3.50)
, (1.3.50)
– для тяжелого компонента
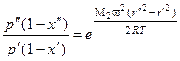 , (1.3.51)
, (1.3.51)
где М1 и М2 – молекулярные массы легкого и тяжелого компонентов; х' и х" – мольные доли легкого компонента на расстоянии от оси r' и r".
Коэффициент разделения α для аппарата вычисляют делением уравнения (1.3.51) на (1.3.50):
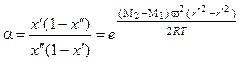 . (1.3.52)
. (1.3.52)
Необходимо отметить, что α зависит от разности молекулярных масс, а не от их отношения, в отличие от диффузионного метода разделения. В этом состоит важное преимущество метода центрифугирования, применяемого для разделения газовых смесей различных компонентов.
В качестве примера, иллюстрирующего порядок величин, рассмотрим разделение бинарной газовой смеси в центрифуге.
Пусть необходимо разделить бинарную газовую смесь изотопов гексафторидов урана 235UF6 и 238UF6).
Вычислим значения соответствующих величин:
ω = 2π·n = 2π·667 = 4190 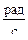 ,
,
где n – число оборотов ротора центрифуги в секунду;
М2 – М1 = = М  – М = 352 – 349 = 3
– М = 352 – 349 = 3 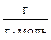 ;
;
r" = 60 мм = 6 см;
r' = 0 мм;
R = 8,314· 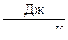 = 8,314·107
= 8,314·107 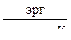 ;
;
T = 300 К.
Из уравнения (1.3.52) определим
α = 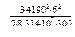 = e0,0127 = 1,01275.
= e0,0127 = 1,01275.
Эта величина примерно соответствует обогащению на одной ступени.
Материальный баланс легкого компонента на единицу высоты легкого потока при r = r' можно выразить в виде
К1 = j 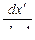 , (1.3.53)
, (1.3.53)
где dx' – вертикальный градиент концентраций легкого компонента в легком потоке, а j – поток.
Если секция обогащения каскада, производящего в секунду р– г-молей продукта с мольной долей легкого компонента хр, состоит из m включенных параллельно центрифуг, то материальный баланс аппарата описывается уравнением:
x' – x" = p·(xp – x")/j·m. (1.3.54)
Оптимальное количество центрифуг, включенных параллельно (mопт), должно соответствовать минимальной общей длине центрифуг,
т. е. величина 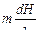 должна быть минимальной. Из этого условия найдем
должна быть минимальной. Из этого условия найдем
mопт = 2p(xp – x)/j(α – 1)·x·(1 – x). (1.3.55)
Причем
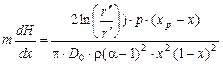 , (1.3.57)
, (1.3.57)
где D0 – коэффициент диффузии, 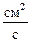 .
.
Полную длину центрифуг ΣН, необходимую в идеальном каскаде для разделения исходной газовой смеси с концентрацией хF на отдельные фракции с концентрациями xW и xp соответственно, получим интегрированием величины в уравнении (1.3.56) в пределах от x F до xp, а соответствующее выражение для секции извлечения – интегрированием в пределах от xW до хF. В результате найдем
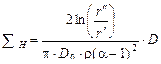 , (1.3.57)
, (1.3.57)
где D – разделительная мощность, определяемая по уравнению
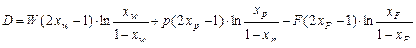 . (1.3.58)
. (1.3.58)
Разделительная мощность имеет ту же размерность, что и потоки. Она является мерой скорости, с которой каскад осуществляет разделение.
Суммарный внутренний (межступенчатый) поток в такой установке составит:
j = 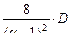 , (1.3.59)
, (1.3.59)
где разделительная мощность
D = 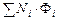 (1.3.60)
(1.3.60)
и
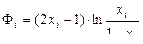 . (1.3.61)
. (1.3.61)
Функция Ф называется разделительным потенциалом. Она безразмерна, зависит только от концентрации х и симметрична относительно точки х = 0,5, в которой, как показано на рис. 1.3.41, она равна нулю.
Суммарную длину каскада центрифуг разделения можно выразить через окружную скорость u = ω·r" и отношение радиусов 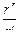 :
:
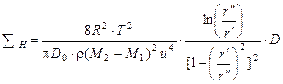 . (1.3.62)
. (1.3.62)
|
Рис. 7.14. Разделительный потенциал
Рис. 7.14. Разделительный потенциал |

|
Рис. 1.3.41 Разделительный потенциал |

Из уравнения (1.3.62) видно, что размеры установки обратно пропорциональны четвертой степени окружной скорости ротора центрифуги. В результате имеем, что 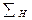 не зависит от абсолютной величины радиусов, но существует оптимальное значение отношения
не зависит от абсолютной величины радиусов, но существует оптимальное значение отношения 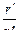 , при котором второй множитель уравнения минимален. Это имеет место при
, при котором второй множитель уравнения минимален. Это имеет место при 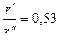 .
.
Центрифуги могут быть надкритическими и подкритическими в зависимости от того, работают ли они выше или ниже резонансной частоты, при которой происходят явления вибрации ротора. Во избежание разрушения центрифуги должны вращаться с частотами, далекими от резонансных, и должны иметь пусковые и тормозные устройства, позволяющие быстро проходить резонансные частоты. Надкритические центрифуги являются более перспективными с технических и экономических позиций.
Для сооружения высокоскоростных центрифуг необходимы особо прочные материалы. Для роторов, выполненных из алюминия или нержавеющей стали, максимальные угловые скорости составляют около 400 м/с. Современные композитные материалы позволяют увеличивать скорость до 700 м/с и выше, что значительно увеличивает эффективность разделения изотопов. В настоящее время разработано и применяется в промышленности большое количество центрифуг, различающихся по диаметру и длине ротора, скоростям вращения и другим инженерным особенностям.
Основное преимущество центрифужного метода заключается в малой энергоемкости, в несколько десятков раз меньшей, чем энергоемкость газодиффузионного метода. Кроме того, для заданной степени обогащения урана требуется существенно меньшее число ступеней по сравнению с газовой диффузией. Поэтому центрифужная технология оказалась более выгодной экономически и начала постепенно вытеснять газодиффузионную технологию.
Центрифужные обогатительные заводы работают в России, Великобритании, Нидерландах, Германии, Японии, Китае. Небольшая установка имеется в Пакистане. Наибольшими мощностями по центрифужному обогащению располагает Россия, которая эксплуатирует четыре завода суммарной производительностью 15 млн. кг ЕРР/год. Центрифужная технология, как уже упоминалось выше, будет внедрена на французском заводе в Трикастене. В США в Портсмуте в 2005 г. планируется ввести в строй первый в стране обогатительный завод на базе центрифужной технологии. На заводе будет установлено 240 центрифуг. Центрифуги разрабатывались более 20 лет в Министерстве Энергетики, их испытания предполагают проводить в Ок-Ридже. Центрифужный завод мощностью 0,1 млн. кг ЕРР/год строится в Бразилии в Резенде.
Таким образом, мировая обогатительная промышленность базируется на двух основных методах: газодиффузионном и центрифужном. Работа над остальными методами либо законсервирована, либо пока не доведена до промышленной реализации. Среди них следует упомянуть следующие методы.
1. Электромагнитный (масс-спектрографический) метод. Разделение основано на различном отклонении ионов разных изотопов в электромагнитном поле. Метод разрабатывался в СССР и США, однако из-за малой производительности, высокой энергоемкости и больших эксплуатационных затрат данная технология не нашла промышленного применения в обогащении урана.
2. Тепловая диффузия. При создании разности температур на противоположных стенках ячейки более легкий 235U диффундирует в сторону горячей зоны быстрее, чем тяжелый 238U. С использованием этой технологии в США был построен опытный завод, который проработал с 1944 по 1945 гг.
3. Химический обмен. Метод базируется на реакциях обмена между химическими соединениями, находящимися в двух несмешивающихся фазах. В результате одна из фаз обогащается 235U. Этот метод лег в основу процесса Chemex (Франция), где несмешивающими фазами являются вода и органическая жидкость, и процесса Asahi (Япония), в котором одна фаза – жидкость, а вторая – твердый адсорбент. Процессы химического обмена широко используются в ядерной промышленности для разделения легких изотопов, например, для получения 10В и тяжелой воды.
4. Газодинамические процессы. Основаны на подаче газового потока с большой скоростью на криволинейную стенку, в результате чего легкие и тяжелые изотопы отклоняются стенкой по-разному. Технологическим газом служит UF6 (~4 %) в водороде или гелии. Разрабатывалось два вида газодинамических систем: с разделительным соплом Бекера (Германия) и с вихревой трубкой (ЮАР). В ЮАР с 1987 по 1995 гг. действовала опытно-промышленная установка производительностью 0,3 млн. кг ЕРР/год.
5. Лазерные методы. Базируются на различии в спектрах поглощения атомов 235U и 238U или молекул, содержащих эти атомы. Лазерное излучение определенной частоты поглощается атомами или молекулами только одного изотопа, переводя их в возбужденное состояние. Для выделения возбужденных атомов или молекул из смеси можно использовать процессы их последующей ионизации, диссоциации или реакции с подходящим химическим соединением. На применении лазерных методов основаны разработки следующих процессов:
– AVLIS (Atomic Vapor Laser Isotope Separation) – США, Россия, Япония;
– SILEX (Separation of Isotopes by Laser Excitation ) – США совместно с Австралией;
– SILVA (Sèparation Isotopique par Laser sur Vapeur Atomique) – Франция;
– MLIS (Molecular Laser Isotope Separation) – США, Россия, Япония, ЮАР;
– CRISLA (Chemical Reaction Isotope Separation by Laser Activation) – США.
В США технология AVLIS была готова к промышленному освоению и к 2005 г. планировалось построить завод производительностью 1 млн. кг ЕРР/год. Однако в 1999 г. работы по программе AVLIS были прекращены из-за финансовых и технических трудностей. Вслед за США Франция отказалась от разработок процесса SILVA, который является аналогом AVLIS.
В табл. 1.3.11 приведено сравнение разных процессов обогащения урана. Анализируя состояние разработок новых методов и тенденций на рынке обогатительных услуг, можно сделать вывод, что в ближайшее время новые процессы вряд ли будут внедряться в промышленное производство. Базовыми промышленными технологиями обогащения урана останутся центрифугирование и газовая диффузия.
Мировые обогатительные мощности в настоящее время, как показано в табл. 1.3.12 составляют около 50 млн. кг ЕРР/год. Учитывая то обстоятельство, что спрос на обогатительные услуги в 2000 г. составлял ~ 35 млн. кг ЕРР/год, а в 2010 г. ожидается на уровне 37-43 млн. кг ЕРР/год, этих производственных мощностей вполне достаточно для обеспечения потребностей ядерной энергетики в ближайшей перспективе. 97 % всех мощностей по обогащению урана принадлежат четырем кампаниям: USEC, Eurodif, Urenco и Tenex.
Цена услуг на обогащение урана по долгосрочным контрактам в 2000 г. была около 80 долл./кг ЕРР. По разным прогнозам, в 2005 г. цены останутся либо на прежнем уровне, либо, в зависимости от обстоятельств, увеличатся до 85-90 долл./кг ЕРР.
3.3.2 Состояние и перспективы разделительных производств
в России
К настоящему времени разделение изотопов урана получило развитие в передовых развитых странах мира, таких, как США, Россия, Франция, Великобритания, Германия, Нидерланды, Япония, Китай.
Более 95 % мощностей мировой разделительной промышленности базируется на двух молекулярных методах: газовой диффузии и центробежном. Имеются полупромышленные установки (Бразилия, ЮАР), работа которых основана на эффекте разделения изотопов в сверхзвуковой струе газа. В США проведены крупномасштабные разработки по лазерному разделению изотопов урана на парах металлического урана, доведенные до создания полупромышленного модуля большой мощности. Однако возникшие технические трудности привели к прекращению работ по этому методу.
3.3.2.1 Развитие разделительной промышленности в СССР
и России
Разделительная промышленность СССР была создана в конце 40-х годов для производства оружейного урана с обогащением по 235U до концентрации 90 %. Первый промышленный завод был создан на основе газодиффузионного метода разделения. В течение короткого исторического периода (1945-1963 гг.) были введены в эксплуатацию четыре газодиффузионных завода, продукция которых шла на изготовление ядерного оружия, а также обеспечение ядерным топливом энергетических установок атомного флота. Часть обогащенного урана направлялась на мирные цели для снабжения ядерным топливом нарождающейся ядерной энергетики. В 60-х гг. велись интенсивные работы по реализации газоцентрифужного метода разделения изотопов урана, и в 1962 г. был пущен в эксплуатацию первый в мире газоцентрифужный завод. Преимущества этого метода, главным образом, его малая энергоемкость (примерно в 20 раз меньше, чем для метода газовой диффузии) позволили отказаться от газовой диффузии и постепенно переоснастить большинство диффузионных корпусов газовыми центрифугами.
Таблица 1.3.11
Сравнение процессов обогащения урана
| Процесс обогащения | Страна | Удельное потребление энергии, кВт·ч/кг ЕРР | Обогащение на одной ступени | Производительность | Технологическое давление | Количество материала, задействованного в процессе одновременно |
| Газовая | США, | 3000 (до 1978 г.) 2500 (после модернизации) | Низкое | Высокая | Умеренное | Большое (десятки тысяч тонн) |
| Франция | 2400 | |||||
| Газовые | СССР-Россия | 120-140 | Высокое | Низкая | Низкое | Малое |
| Великобритания, Нидерланды, Германия (фирма Urenco) | 100, новые модели 50 | |||||
| Газодинамические процессы: сопло вихревая трубка | Германия ЮАР | 3300 3500 | Среднее | Средняя | Умеренное | Малое |
| Химический обмен: Chemex-процесс | Франция | 600 | Среднее | Высокая | Низкое | Большое |
| Лазерный метод: AVLIS | США | 100-130 | Очень высокое | Очень низкая | Очень низкое | Малое |
Таблица 1.3.12
Мощности заводов по обогащению урана по состоянию на 2003 г.
| Страна | Местоположение завода | Организация, владеющая или управляющая заводом | Мощность, млн. кг ЕРР/год |
|
Газодиффузионная технология | |||
| США | Падука, шт. Кентукки | USEC | 11,3 |
| Портсмут, шт. Огайо (закрылся в 2001 г.) | 7,4 | ||
| Франция | Трикастен | Eurodif | 10,8 |
| Китай | Ланчжоу | CNNC | 0,9 |
| Всего (газовая диффузия) | 30,4 | ||
|
Центрифужная технология | |||
| Россия | Новоуральск | Минатом (Tenex) | 7,0 |
| Северск | 4,0 | ||
| Зеленогорск | 3,0 | ||
| Ангарск | 1,0 | ||
| Великобритания | Капенхерст | Urenco | 2,44 |
| Нидерланды | Алмело | 1,95 | |
| Германия | Гронау | 1,46 | |
| Япония | Рокасё | JNFL | 1,05 |
| Нингайо Тоге | JNC | 0,2 | |
| Китай | Ланчжоу | CNNC | 0,5 |
| Ханжонг | 0,5 | ||
| Пакистан | Кахута | PAEC | 0,005 |
| Всего (центрифугирование) | 23,1 | ||
| Суммарная мощность всех заводов | 53,5 | ||
В настоящее время Россия обладает самыми большими разделительными мощностями газоцентрифужных заводов в мире, что позволяет ей успешно конкурировать на международном рынке обогащенного урана, эффективно работать в изменившихся экономических условиях и использовать ранее накопленные богатые отвалы как альтернативу дефицита природного урана. За прошедшие годы были разработаны и пущены в серийное производство семь поколений газовых центрифуг, из которых последние три эксплуатируются по настоящее время.
3.3.2.2 Современное состояние и оборудование разделительных заводов
В общем объеме установленных на предприятиях России газовые центрифуги 5-го поколения составляют около 48 %, 6-го – 49 %, 7-го – 3%. В настоящее время на предприятиях проводится плановая замена отработавших ресурс газовых центрифуг 5-го поколения на центрифуги 7-го поколения. К 2010 г. газовые центрифуги 5-го поколения исчерпают ресурс, и в дальнейшем потребуется заменять выработавшие ресурс газовые центрифуги 6-го поколения.
Для производства центрифуг используются высокопрочные алюминиевые сплавы, высокоточные штамповки и трубные заготовки, упрочняющие волокна для роторных деталей (армос, стекловолокно), ужестчающие угольные композиции для ротора, искусственные лейкосапфиры для опорных подшипников, специальное литье из цветных металлов. Осуществляется широкая кооперация предприятий, изготавливающих исходные материалы и комплектующие изделия.
На предприятиях, изготавливающих газовые центрифуги, разработаны и внедрены специальные высокоточные станки для механической обработки роторных деталей, специальные намоточные программируемые станки, печи для полимеризации обмотки роторов, специальные сборочные конвейеры роторов и агрегатов, серия обкаточных и испытательных стендов.
На разделительных газоцентрифужных заводах используются высокоразвитая двухуровневая автоматизированная система управления технологическим процессом с контролем нескольких миллионов параметров как глобальных по всему заводу, так и по каждой центрифуге, процессом в аварийных ситуациях и при полном отключении внешнего электроснабжения. На заводах используются специальные датчики контроля параметров процесса.
Для электроснабжения двигателей газовых центрифуг применяются специализированные статические преобразователи, вырабатывающие электропитание повышенной частоты с высокой степенью ее стабилизации, не зависящие от колебаний напряжения и частоты во внешней электросети. В корпусах газоцентрифужных заводов, суммарный объем которых превышает несколько миллионов м3, постоянно поддерживается температура 18-20 °С. Современные газоцентрифужные заводы представляют собой полностью автоматизированные предприятия, работающие круглосуточно в течение ресурсного срока, достигающего для последних поколений газовых центрифуг 25-30 лет.
Технологический процесс разделения осуществляется при низком давлении (5-40 мм рт. ст., абс.), что обеспечивает отсутствие каких-либо выделений радиоактивных и химически вредных веществ в рабочие помещения, поддержание выбросов в окружающую среду на уровне ниже санитарных норм.
Производство гексафторида урана из высших оксидов урана
Дата: 2019-02-25, просмотров: 412.