Одним из наиболее распространенным методом ускорения химических реакций является катализ. Этот метод заключается в том, что в реакционную систему добавляют катализаторы - вещества, резко увеличивающие скорость реакции, но не расходующиеся в результате ее протекания. Замедление реакции осуществляется при помощи ингибиторов (отрицательных катализаторов).
Различают катализ гомогенный и гетерогенный. Примером гомогенной реакции является реакция нейтрализация в растворе: НС1+КОН=KС1+Н2О. Примером гетерогенной реакции является термическое разложение карбоната кальция: СаСОз = СаО+СO2.
Скорость гетерогенной реакции зависит от площади контактирующих поверхностей. Если катализатор и взаимодействующие вещества находятся в одной фазе, катализ называется гомогенным. Если катализатор находится в системе в виде самостоятельной фазы - гетерогенным.
Влияние катализатора на скорость реакции при гомогенном катализе объясняется образованием промежуточного соединения катализатора с реагирующим веществом. Так, медленно протекающая реакция А+В=АВ ускоряется катализатором К, вследствие образования реакционноспособного промежуточного соединения А+К=АК, которое взаимодействует с другим исходным веществом В с образованием продукта реакции АВ и выделением катализатора К в первоначальном виде АК+В=АВ+К.
Примером гомогенного катализа может служить окисление диоксида серы в серную кислоту при использовании в качестве катализатора N O 2 :
2 S О2+О2+2Н2О=2Н2 S О4.
Эта реакция лежит в основе одного из промышленных методов получения серной кислоты.
Химическое равновесие. Константа химического равновесия.
Реакции, которые при заданных условиях протекают одновременно в двух взаимопротивоположных направлениях, называются обратимыми и обозначаются m А+ n В↔рС+ dD . Согласно закону действия масс, скорости прямой и обратной реакций соответственно равны: ύ1 = k 1 [ A ] m [ B ] n , ύ2 = k 2 [ C ] p [ D ] d
При достижении равновесия скорость прямого процесса становится равной скорости обратного: ύ= ύ2. Следовательно, k 1 [ A ] m [ B ] n = k 2 [ C ] p [ D ] d , обозначив Kp = k 1 / k 2 , получим:
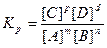 , где Кр - константа равновесия.
, где Кр - константа равновесия.
Значение величины Кр дает возможность судить о преимуществе того или иного процесса. Для гетерогенных реакций в выражение для константы равновесия входят концентрации только тех веществ, которые находятся только в газовой или жидкой фазах. Например, для реакции СО2(г)+С(тв)↔2СО(г) КР=[СО]2/[СО]. Константа равновесия зависит только от температуры и природы реагентов.
Изменение энергии Гиббса Δ G обратимого процесса связано с константой химического равновесия следующим уравнением: Δ G = – R Т1 n Кр. Здесь R - универсальная газовая постоянная, Т- абсолютная температура.
Эта зависимость позволяет, зная ΔG, вычислить константу равновесия и равновесные концентрации реагирующих веществ. Переходя к десятичному логарифму и подставляя значение R, уравнение принимает вид ΔG =-5,71lgКР, кДж/моль.
В условиях химического равновесия концентрации (или парциальные давления в случае газов) исходных веществ и продуктов реакции не изменяются во времени и называются равновесными концентрациями (или парциальными давлениями). Они обозначаются так же, но с индексом р: [СО2]Р, [СО]Р.
Принцип Ле-Шателье.
Химическое равновесие, отвечающее равенству скоростей прямой и обратной реакций, и минимальному значению энергии Гиббса, является наиболее устойчивым состоянием системы при заданных условиях, и остается неизменным до тех пор, пока сохраняются постоянными параметры, при которых равновесие установилось. При изменении условий равновесие нарушается и смещается в ту или иную сторону. Направленные смещения равновесия определяются принципом Ле-Шателье: если на систему, находящуюся в равновесии, оказать внешнее воздействие, то равновесие смещается в том направлении, которое ослабляет эффект внешнего воздействия.
Смещение равновесия может быть вызвано изменением температуры, давления, концентрации одного (или нескольких) реагента.
При повышении температуры увеличивается константа равновесия эндотермического процесса (ΔН>0). Это значит, что при повышении температуры равновесие смещается вправо, в сторону прямой реакции, идущей с поглощением теплоты. Константа равновесия экзотермического процесса (ΔН<0) при повышении температуры уменьшается. Это значит, что при повышении температуры равновесие экзотермической реакции смещается влево.
Смещение равновесия может быть вызвано изменением концентрации одного из компонентов: добавлением вещества в равновесную систему или выводом его из системы. Согласно принципу Ле-Шателье, при увеличении концентрации одного из исходных веществ равновесие смещается в правую сторону, а при увеличении концентрации одного из продуктов реакции - в левую.
Если в обратимой реакции участвует хотя бы одно газообразное вещество, смещение равновесие может быть вызвано изменением давления. Повышение давления при постоянной температуре равносильно сжатию газа, то есть увеличению его концентрации. При увеличении концентрации газообразного компонента скорость реакции в соответствии с законом действия масс возрастает, что приводит к смещению равновесия в направлении уменьшения концентрации газообразного компонента. При понижении давления при постоянной температуре газ расширяется, и его концентрация в системе падает. Это вызывает уменьшение скорости реакции, равновесие смещается в направлении увеличения давления газа. Например, при увеличении давления в системе СаСОз ↔ СаО+СO2 возрастает скорость обратной реакции ύ = k2[СО2], что приводит к смещению равновесия в левую сторону. При понижении давления на ту же систему скорость обратной реакции уменьшается, это приводит к смещению равновесия в правую сторону. В системе 2НС1↔Н2+Cl2 все вещества газообразные, при изменении давления смещения не произойдет, так как обе скорости возрастут одинаково.
Итак, в соответствии с принципом Ле-Шателье при повышении давления равновесие смещается в сторону образования меньшего числа молей газообразных веществ и соответственно в сторону уменьшения давления в системе. Наоборот, при внешнем воздействии, вызывающим понижение давления, равновесие смещается в сторону образования большего числа молей газа, что противодействует внешнему воздействию и вызывает увеличение давления в системе.
Тема: «Растворы. Растворы неэлектролитов»
План
1. Общая характеристика растворов. Растворимость
2. Способы выражения концентрации растворов
3. Общие свойства растворов. Законы Рауля и Вант-Гоффа.
Растворение - сложный физико-химический процесс. В зависимости от природы растворенного вещества и растворителя преобладает либо физическая, либо химическая сторона явления. Физическая - статистическое распределение растворенного вещества (разрушение кристаллической решетки, процесс эндотермический, ΔН1>0). Химическая - гидратация образующихся частиц, процесс экзотермический, ΔН2<0. Знак суммарного процесса растворения определяется соотношением величин ΔН1 и ΔН2.
Очень многие химические реакции, в том числе технические и жизненно важные, протекают в жидких растворах. Растворами называют гомогенные смеси переменного состава, находящиеся в состоянии химического равновесия. Растворы - это как минимум двухкомпонентные системы. Растворитель сохраняет свое фазовое состояние при образовании раствора. Его концентрация по определению выше, чем концентрации других компонентов.
Многие свойства растворов зависят от их концентрации. Концентрацией называется отношение количества или массы вещества, содержащегося в системе, к объему или массе этой системы.
Молярная концентрация вещества В Св – отношение количества вещества ( 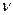 , моль) к объему системы (V, л):
, моль) к объему системы (V, л): 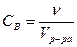 моль/л или моль/м3.
моль/л или моль/м3.
Молярная доля вещества В (хв) - отношение количества вещества (моль), содержащегося в системе, к общему количества вещества (моль). Молярная доля может быть выражена в долях единицы и в процентах.
Объемная доля вещества В (φв) - отношение объема компонента, содержащегося в системе, к общему объему системы. Объемная доля может быть выражена в долях единицы и в процентах.
Массовая доля вещества В (ωв) - отношение массы данного компонента к общей массе этой системы. Может быть выражена в долях единицы и в процентах.
Растворимость. При растворении веществ возникает равновесие, при котором скорость растворения фазы равна скорости ее образования.
Растворенное вещество (фаза) ↔ раствор
При равновесии изменение энергии Гиббса равно нулю. Раствор, в котором устанавливается равновесие между растворением и образованием (осаждением, кристаллизацией, выделением вещества), называется насыщенным, а концентрация такого раствора — растворимостью. Растворимость газов уменьшается с увеличением температуры. Растворимость твердых веществ может как увеличиваться, так и уменьшаться, а также мало зависеть от температуры.
Растворимость вещества зависит от свойств растворителя. Вещества с одинаковым характером связей имеют большую тенденцию к взаимной растворимости - подобное растворяется в подобном. Например, полярные и ионные вещества хорошо растворимы в полярных растворителях, неполярные - в неполярных растворителях.
Общие свойства растворов - те свойства, которые не зависят от природы растворенных веществ, а только от их концентрации. Они также называются коллигативными. Такие свойства характерны для идеальных растворов.
Идеальным называют раствор, в котором не происходит химической реакции между компонентами, а силы межмолекулярного взаимодействия между компонентами одинаковы. Энтальпия образования такого раствора равна нулю, каждый компонент является независимым от других. К идеальным растворам по своим свойствам приближаются лишь очень разбавленные растворы.
К общим свойствам растворов относят понижение давления насыщенного пара растворителя над раствором и температуры замерзания, повышение температуры кипения и осмотическое давление. Растворенным веществом в таких растворах является нелетучее вещество, давлением пара которого можно пренебречь.
Закон Рауля. Понижение давления насыщенного пара растворителя А над раствором ΔрА пропорционально молярной доле растворенного нелетучего вещества
хв: р°А – рА = Δр а = р°А хВ,
где р°а, ра - давления насыщенного пара растворителя соответственно над чистым растворителем и раствором. Из уравнения видно, что с увеличением содержания нелетучего растворенного вещества давление пара растворителя над раствором уменьшается, то есть р°а>ра.
Из закона Рауля следует:
1. Температура кипения раствора выше температуры кипения растворителя, потому что давление насыщенного пара растворителя над раствором становится равным атмосферному (условие кипения жидкости) при более высокой температуре, чем в случае чистого растворителя. Повышение температуры кипения пропорционально моляльности раствора:
ΔТкип=КэСm.
Здесь Кэ - эбуллиоскопическая константа растворителя.
2. Температура замерзания (кристаллизации) раствора ниже температуры замерзания чистого растворителя, потому что давление пара растворителя над раствором ниже, чем над растворителем. Понижение температуры замерзания пропорционально моляльности раствора:
ΔТ3ам=КкСm.
Здесь Кк - криоскопическая константа.
Значения эбуллиоскопической и криоскопической констант зависят от природы растворителя; для воды они составляют соответственно 0,52 и 1, 85 кг·К/моль.
Используя следствие из закона Рауля, можно определить молярную массу вещества. Для этого экспериментально определяют изменение температуры кипения или замерзания раствора. Зная массу растворенного вещества mв, его молярную массу находят по уравнению:
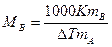 ,
,
где К - либо эбулио-, либо криоскопическая константы.
Закон Вант-Гоффа. Самопроизвольный переход растворителя через полупроницаемую мембрану, разделяющую раствор и растворитель, либо два раствора с различной концентрацией растворенного вещества, называется осмосом. Перегородка пропускает только молекулы растворителя, которые переходят из раствора с меньшей концентрацией в более концентрированный раствор. Концентрированный раствор разбавляется и увеличивается высота его столба. Количественно осмос характеризуют осмотическим давлением, равным силе, приходящейся на единицу площади поверхности и заставляющей молекулы растворителя проникать через полупроницаемую перегородку. Осмотическое давление возрастает с увеличением концентрации растворенного вещества и температуры. Вант-Гофф предложил для осмотического давления применить уравнение состояния идеального газа pV=nRТ, или p=nRТ/V. Отсюда
p=СRТ - осмотическое давление.
С -молярная концентрация раствора.
Осмос играет важную роль в биологических процессах, обеспечивая поступление воды в клетки и другие структуры. Растворы с одинаковым осмотическим давлением называются изотоническими; если осмотическое давление выше внутриклеточного, то оно называется гипертоническим, ниже -гипотоническим. Среднее осмотическое давление крови при 36°С равно 78 кПа. Гипертонические растворы, например, соли и сахара (рассол), широко применяют для консервирования овощей (огурцов), так как они вызывают удаление воды из микроорганизмов. Огурцы в крепком рассоле сморщиваются, в гипотоническом растворе - разбухают.
Законы Рауля и Вант-Гоффа соблюдаются лишь в разбавленных растворах неэлектролитов, являющихся моделью идеальных растворов, в реальных растворах вместо концентрации используется активность, о которой подробно будет сказано в следующей лекции.
Тема: «Растворы электролитов»
План
1. Растворы электролитов. Основные положения теории электролитической диссоциации.
2. Степень электролитической диссоциации.
3. Сильные и слабые электролиты. Константа диссоциации.
4. Активность электролитов. Применение законов Рауля и Вант-Гоффа к растворам электролитов.
5. Диссоциация воды. Ионное произведение воды. Водородный показатель.
6. Ионно-молекулярные реакции в растворах. Произведение растворимости.
7. Реакции нейтрализации и гидролиз солей.
Общие свойства растворов электролитов
Вещества, растворы или расплавы которых проводят электрический ток, называются электролитами. Свойства электролитов были рассмотрены и обобщены в трудах Аррениуса, развиты в трудах И.А. Каблукова, В.А. Кистяковского, на основе гидратной теории растворения Д.И. Менделеева. Основные положения теории электролитической диссоциации (ионизации):
1. При растворении солей, кислот и оснований в воде происходит диссоциация (ионизация) этих соединений с образованием электрически заряженных частиц -катионов и анионов.
2. Электрическая проводимость растворов солей, кислот и оснований в воде прямо пропорциональна общей концентрации ионов в растворе.
Уравнение электролитической диссоциации можно записать, опустив промежуточные стадии, указав лишь начальные и конечные продукты реакции:
АВ+(n+m)Н2О ↔ АР+nН2О + Вq–mН2О (1)
Коэффициенты n и m меняются с изменением концентрации, температуры и других параметров раствора. Поэтому молекулы растворителя обычно опускают и записывают: АВ ↔Ap+ + Вq–.
Растворами электролитов являются растворы щелочей, солей и неорганических кислот в воде и других полярных растворителях (жидком аммиаке, ацетонитриле и др.). Растворы электролитов являются ионными проводниками (проводниками второго рода).
В растворах электролитов наблюдаются отклонения от законов Вант-Гоффа и Рауля. По закону Рауля при растворении 0,1 моль неэлектролита (глюкозы) в 1000 г воды температура замерзания снижается на 0,186 К. Поэтому Вант-Гофф ввел в уравнение p=СRТ поправочный коэффициент i, который назван изотоническим коэффициентом. Для любых разбавленных растворов Росм=iСRТ. Изотонический коэффициент характеризует отклонение от законов идеальных растворов вследствие электролитической диссоциации.
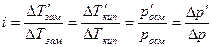
Здесь 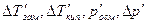 – изменения температур замерзания и кипения, осмотического давления и понижения давления пара для электролита к аналогичному свойству раствора неэлектролита той же концентрации (
– изменения температур замерзания и кипения, осмотического давления и понижения давления пара для электролита к аналогичному свойству раствора неэлектролита той же концентрации ( 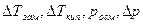 ). Для раствора электролита всегда i>1, для раствора неэлектролита i=1.
). Для раствора электролита всегда i>1, для раствора неэлектролита i=1.
Степень диссоциации электролитов.
Отношение числа молекул, диссоциированных на ионы, к общему числу молекул растворенного электролита называется степенью диссоциации (ионизации) ά: ά = n/nо, где n - число частиц, подвергшихся диссоциации, nо - общее число растворенных частиц. По степени диссоциации электролиты делятся на слабые (ά <3%) и сильные (ά >30%). Степень диссоциации зависит от природы (полярности) растворителя. Чем более полярна молекула растворителя, тем при прочих равных условиях выше степень ионизации растворенного вещества. Поскольку электролитическая диссоциация сопровождается тепловым эффектом, то ά зависит от температуры, причем ее влияние можно оценить по принципу Ле-Шателье: если диссоциация представляет собой эндотермический процесс, то с повышением температуры степень диссоциации растет, с понижением температуры уменьшается.
Сильно влияет на степень электролитической диссоциации концентрация раствора. Если рассматривать диссоциацию как равновесный обратимый химический процесс, то в соответствии с принципом смещения равновесия разбавление водой увеличивает количество диссоциированных молекул, то есть степень диссоциации с разбавлением растет.
Константа диссоциации
Процесс электролитической диссоциации удобнее характеризовать константой диссоциации, применив к нему законы химического равновесия. Так, для реакции КА↔К++А– константа диссоциации 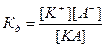 . Здесь и далее в квадратных скобках обозначаются молярные концентрации компонентов. Чем больше
. Здесь и далее в квадратных скобках обозначаются молярные концентрации компонентов. Чем больше
величина 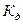 , тем сильнее электролит. Константа диссоциации зависит от природы диссоциирующего вещества и растворителя, а также от температуры, и не зависит от концентрации раствора. Между константой и степенью диссоциации существует количественная зависимость. Пусть в рассмотренном выше процессе диссоциации общее количество растворенного вещества КА равно С, а степень диссоциации ά. Тогда [К+]=[А-]= άС, и, соответственно, концентрация недиссоциированных частиц [КА]= (1- ά)С. Подставив это значение в выражение для константы диссоциации, получим:
, тем сильнее электролит. Константа диссоциации зависит от природы диссоциирующего вещества и растворителя, а также от температуры, и не зависит от концентрации раствора. Между константой и степенью диссоциации существует количественная зависимость. Пусть в рассмотренном выше процессе диссоциации общее количество растворенного вещества КА равно С, а степень диссоциации ά. Тогда [К+]=[А-]= άС, и, соответственно, концентрация недиссоциированных частиц [КА]= (1- ά)С. Подставив это значение в выражение для константы диссоциации, получим:
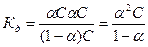 .
.
Это соотношение называется законом разбавления Оствальда. Для слабых электролитов, когда ά много меньше 1, 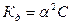 . Отсюда
. Отсюда
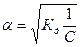 , где V=1/С - разбавление.
, где V=1/С - разбавление.
Из уравнения Оствальда следует, что ά уменьшается с увеличением концентрации слабого электролита. Например, при разбавлении раствора в 1 00 раз степень диссоциации возрастает в 10 раз.
В растворах слабых электролитов взаимодействие ионов друг с другом относительно невелико, вследствие их незначительной концентрации. Сильные электролиты в растворах диссоциированы практически полностью, поэтому концентрация их ионов велика и свойства таких растворов зависят от степени взаимодействия образующихся ионов друг с другом и с полярными молекулами растворителя. За счет этого взаимодействия создается впечатление, что диссоциация прошла и в растворе имеется некоторое количество недиссоциированных молекул. Поэтому ά, определяемая в растворах сильных электролитов экспериментально, является кажущейся и ее значение менее 100%. Поэтому для растворов сильных электролитов не применимы законы идеальных растворов. Чтобы эти законы использовать для описания реальных растворов, Льюис ввел представление об эффективной концентрации - активности а: а=γС, где С - концентрация, γ - коэффициент активности (величина безразмерная). Активность и концентрация измеряются в одних и тех же единицах. Коэффициент активности характеризует степень отклонения свойств данного раствора от свойств идеального. Для бесконечно разбавленных растворов электролитов, где практически отсутствует взаимодействие ионов, а=С и γ=1. Если вместо С в уравнения Вант-Гоффа, Рауля, Оствальда подставлять экспериментальные значения а, то уравнения остаются справедливыми и для сильных электролитов, и вообще для любых реальных растворов.
Вода служит не только наиболее распространенным растворителем, но и сама является идеальным амфолитом. Процесс диссоциации воды называется самоионизацией или автопротолизом, и может быть записан: Н2О+Н2О↔Н3О++ОН–. Часто этот процесс записывают в более простом виде: Н2О↔Н++ОН–. Константа диссоциации этого процесса 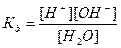 , более строго через активности ионов:
, более строго через активности ионов: 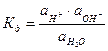 .
.
Так как вода - слабый электролит, можно использовать данные выражения с учетом того, что концентрация воды постоянна: [Н2О]=55,56 моль/л. Тогда [Н+][ОН-]=55,56 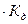 =КW=10-14 – ионное произведение воды. Так как при диссоциации воды [Н+]=[ОН-]=
=КW=10-14 – ионное произведение воды. Так как при диссоциации воды [Н+]=[ОН-]= 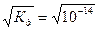 =10-7 моль/л.
=10-7 моль/л.
Ионы водорода являются носителями кислотных свойств, гидроксил-анионы - щелочных. Поэтому при равенстве этих ионов раствор будет нейтральным. Для характеристики кислотности (щелочности) среды введен водородный показатель рН=-lg[Н+]. При рН=7 среда нейтральная, для кислых растворов рН меньше 7, для щелочных рН больше 7.
Аналогичным образом реакция среды может быть охарактеризована гидроксильным показателем рОН=-lg[ОН–]. Для воды рН=рОН=7, рН+рОН=14.
Рассмотрим обменную реакцию КС1+АgNОз=АgС1+КNО3. Равновесие в такой реакции смещено вправо, так как хлорид серебра - малорастворимое соединение. Для насыщенного раствора АgС1, находящегося в равновесии с твердой фазой (осадком), будет характерен следующий процесс: АgCl↔Аg++С1–. Константа равновесия этого гетерогенного процесса Кр=Пр= [Аg+][С1–]. Она не зависит от концентрации твердой фазы (АgС1), является постоянной величиной при данной температуре. Эту величину называют произведением растворимости Пр. Более строго в Пр используют не концентрации, а активности ионов электролита Пр= 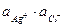 . Применение понятия о Пр аналогично использованию КW. КW определяет ионное равновесие для малодиссоциированного слабого электролита, а произведение растворимости описывает описывает аналогичный процесс для сильного, но малорастворимого соединения.
. Применение понятия о Пр аналогично использованию КW. КW определяет ионное равновесие для малодиссоциированного слабого электролита, а произведение растворимости описывает описывает аналогичный процесс для сильного, но малорастворимого соединения.
Реакции нейтрализации и гидролиз солей
К обменным реакциям в растворах относят взаимодействие между кислотами и основаниями, в результате которых образуется соль и вода. Такие реакции называются реакциями нейтрализации и доходят до конца только тогда, когда единственным малодиссоциированным веществом в системе является вода, например: НС1 + КОН = КС1 + Н2О. Сокращенное ионное уравнение этого процесса Н++ОН–=Н2О. Оно показывает, что равновесие полностью смещено в сторону образования воды. Взаимная нейтрализация кислот и оснований, отличающихся друг от друга по силе, до конца не протекает. Так, при взаимодействии слабой уксусной кислоты с гидроксидом калия в системе из четырех веществ два слабодиссоциированы: СНзСООН + КОН ↔ Н2О + СНзСООК, или в сокращенном ионном виде СНзСООН + ОН– ↔ Н2О + СНзСОО–. Эта реакция обратима. Обратная реакция - взаимодействие соли и воды - называется реакцией гидролиза. Гидролизу подвергаются соли, образованные:
1) Слабым основанием и сильной кислотой;
2) сильным основанием и слабой кислотой;
3) слабыми основанием и кислотой.
Соли, образованные сильным основанием и сильной кислотой, гидролизу не подвергаются.
Приведем примеры реакций гидролиза.
Гидролиз соли, образованной сильным основанием и слабой кислотой - ацетата натрия. Соль в растворе полностью диссоциирует: СНзСООNa ↔ СНзСОО– + H+.
Гидролиз в ионной форме: СН3СОО-+Н2О↔СН3СООН+ОН-.
В результате гидролиза реакция среды стала основной (избыток ОН-), рН>7. Показателем глубины протекания гидролиза является степень гидролиза β:
β=Сгидр/С,
где Сгидр – концентрация гидролизованных молекул, С – исходная концентрация растворенных молекул соли. Гидролиз – процесс эндотермический, поэтому, в соответствии с принципом Ле-Шателье, с повышением температуры степень гидролиза увеличивается.
Принимая, что в разбавленных растворах активность ионов мало отличается от их концентрации (С=а), можно записать константу равновесия 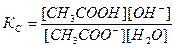 . Так как концентрация воды при гидролизе изменяется мало, принимаем ее постоянной, и, Кс[Н2О]=КГ, получаем константу гидролиза:
. Так как концентрация воды при гидролизе изменяется мало, принимаем ее постоянной, и, Кс[Н2О]=КГ, получаем константу гидролиза: 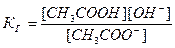 . Подставляя вместо [ОН-]=Kw/[H+], получаем:
. Подставляя вместо [ОН-]=Kw/[H+], получаем: 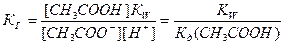 .
.
Отсюда следует, что гидролиз идет тем глубже, чем ниже константа диссоциации слабого электролита, образующегося при гидролизе.
Гидролиз солей, образованных сильной кислотой и слабым основанием, например, хлорида аммония:
NH4Cl↔NH4+ + Cl- (диссоциация соли)
NH4+ + H2O ↔ NH4OH + H+, среда кислая, рН<7.
Степень гидролиза и константа гидролиза для данной реакции описываются аналогично приведенным для реакции гидролиза соли, образованной слабой кислотой и сильным основанием.
Гидролиз соли, образованной слабой кислотой и слабым основанием, протекает как по катиону, так и по аниону. Константа гидролиза зависит от констант диссоциации слабых кислоты НА и основания ВОН: 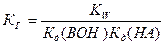 . В зависимости от силы кислоты и основания реакция среды может быть как щелочной, так и кислой.
. В зависимости от силы кислоты и основания реакция среды может быть как щелочной, так и кислой.
Итак, гидролиз изменяет реакцию среды (подкисляет или подщелачивает). Степень гидролиза возрастает с разбавлением раствора и при увеличении температуры.
ЛЕКЦИЯ ПО ТЕМЕ: «ДИСПЕРСНЫЕ СИСТЕМЫ»
1. Понятия о дисперсных системах, их классификация.
2. Коллоидные растворы.
3. Строение и заряд коллоидной частицы. Мицелла
4. Оптические свойства коллоидных растворов
5. Электрические свойства коллоидных растворов
6. Коагуляция коллоидных растворов
7. Коллоидные растворы в природе и технике
Все дисперсные системы состоят из сплошной фазы, называемой дисперсионной средой, и прерывистой фазы (частиц), называемой дисперсной фазой.
От линейных размеров частиц дисперсной фазы зависит гомогенность или гетерогенность дисперсной системы. Гомогенные дисперсные системы называются истинными растворами, или просто растворами. Истинные растворы содержат молекулы или атомы, размеры которых не превышают 5 нм (5˙10-9 м).
Гетерогенные дисперсные системы подразделяют на: а) грубодисперсные системы, у которых частицы имеют размер 1000 нм (10-6 м) и более; б) коллоидные системы, размер частиц которых лежит в пределах от 1 до 500 нм (10-9 до 5˙10-7 м).
К грубодисперсным системам относятся суспензии, эмульсии, пены.
Суспензии представляют собой системы, состоящие из раздробленного твердого вещества и жидкости, в которой распределена дисперсная фаза. Например, крахмал в холодной воде, шоколад (какао в масле), мутная вода. Примеры концентрированных суспензий – пасты, взвесь глины в воде.
Эмульсии – образуются двумя несмешивающимися жидкостями; обычно одной из фаз является вода. Примеры эмульсий: молоко, майонез, маргарин, бензол в воде.
Пены – грубодисперсные системы, состоящие из ячеек, заполненных газом, и отделенных друг от друга пленками очень малой толщины. К пенам относятся: мыльная пена, мусс, пелус, пенопласты.
Характерным признаком грубодисперсных систем является то, что частицы дисперсной фазы видны в обычный микроскоп или даже невооруженным глазом.
Частицы в коллоидных системах уже невозможно различить в обычный микроскоп, так как их размеры меньше длины волны видимого света.
Дисперсные системы с частицами коллоидных размеров называют золями. По характеру дисперсионной среды различают: гидрозоли (растворитель – вода), аэрозоли (мельчайшие капельки жидкости, тонкораспыленные в газе, например, туман), мелкие твердые частицы в газе (дым).
Помимо классификации дисперсных систем по размерам частиц, существует и другая классификация – по агрегатному состоянию дисперсной фазы и дисперсионной среды.
| Дисперсная фаза | Дисперсионная среда | Название системы |
| Жидкость Твердое тело Газ Жидкость Твердое тело Газ Жидкость Твердое тело | Газ Газ Жидкость Жидкость Жидкость Твердое тело Твердое тело Твердое тело | Аэрозоли (туман, дым, пыль) Пены Эмульсии Суспензии Твердые пены Твердые эмульсии Сплавы, тв.золи |
Коллоидные системы
Коллоидные растворы отличаются от истинных рядом свойств:
1. Малой скоростью перемещения ввиду малой скорости диффузии;
2. Повышенной вязкостью;
3. Гетерогенностью.
От грубодисперсных систем они отличаются относительной устойчивостью. Для коллоидов характерна очень развитая поверхность, в связи с чем в коллоидных растворах исключительно важную роль играет адсорбция.
Существует два основных пути получения коллоидных растворов.
1. Дисперсионный – измельчение частиц грубодисперсных систем до размеров, соответствующих коллоидам. При этом используются обычно физические методы с применением различных диспергаторов и коллоидных мельниц.
2. Конденсационный – укрупнение частиц истинных растворов (ионов, молекул) путем их ассоциации до размеров, соответствующих коллоидам. Применяются обычно химические методы: осаждение, гидролиз, окисления-восстановления, нейтрализации.
Например, в результате гидролиза солей железа (III) получают его гидроксид: Fe3+ + 3H2O = Fe(OH)3 + 3H+.
Для повышения устойчивости коллоидов в раствор вводят стабилизаторы (ПАВ).
Строение и заряд коллоидной частицы. Мицелла
Коллоидная частица состоит из ядра, адсорбирующего из окружающей среды ионы одного вида. Эти ионы называют зарядообразующими, их химическая природа близка химической природе ядра коллоидной частицы.
Например,
| золь | Ядро коллоидной частицы | Зарядообразующие ионы |
| AgJ | mAgJ | Ag+ или J- |
| Fe(OH)3 | mFe(OH)3 | Fe3+, Fe(OH)2+ Или Fe(OH)2+ |
Ядро коллоидной частицы с адсорбированными зарядообразующими ионами притягивает к себе из среды ионы противоположного знака (заряда). Зарядообразующие ионы и противоионы гидратированы. Весь этот комплекс и называют коллоидной частицей. В состав коллоидной частицы входит только часть имеющихся в системе противоионов – их называют связанными. Другая часть противоионов остается в дисперсной среде (жидкой фазе). Эти противоионы называют свободными. Они определяют заряд дисперсионной среды. Все сочетание, состоящее из коллоидной частицы и дисперсионной среды (гидратированных свободных противоионов) называют мицеллой. Мицелла – это структурная единица коллоидного раствора.
Примерный состав коллоидных частиц и мицелл золей иодида серебра и гидроксида железа (III)
| золь | Коллоидная частица | мицелла |
| AgJ | [(mAgJ)nAg+(n-x)NO3-yH2O]x+ | {[(mAgJ)nAg+(n-x)NO3-yH2O]x++xNO3-zH2O}0 |
| Fe(OH)3 | [mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ | {[mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ + 3xCl-zH2O}0 |
Мицелла имеет сложное строение. Пример строения мицеллы Fe(OH)3 приведен ниже.
[mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ + 3xCl-
Ядро связанные противоионы свободные противоионы
Адсорбционный слой диффузионный слой
 Коллоидная частица
Коллоидная частица
 Мицелла
Мицелла
Суммарно заряд частицы равен разности зарядов адсорбированных ионов и противоионов. Вокруг частицы находится диффузионный слой ионов, заряд которых равен заряду коллоидной частицы. Коллоидная частица и диффузионный слой образуют электронейтральную мицеллу.
Оптические свойства коллоидных растворов
При прохождении через дисперсную систему свет может поглощаться, отражаться или рассеиваться частицами. Поглощение света – явление избирательное. Одни вещества полностью поглощают свет, другие поглощают только лучи определенной части спектра. Отражение света поверхностью частиц возможно только в грубодисперсных системах. Отражение света проявляется в мутности таких дисперсных систем как в проходящем свете, так и при боковом их освещении.
Для типичных коллоидных систем наиболее характерным оптическим свойством является рассеивание света по всем направлениям. Размеры коллоидных частиц меньше длины световой волны, и поэтому рассеивание света обусловлено не отражением его от поверхности частиц, а дифракцией. Рассеивание света при освещении коллоидного раствора было исследовано Тиндалем. Путь света виден при наблюдении сбоку в виде светящейся полосы (конус Тиндаля). Это свечение было названо опалесценцией.
Электрические свойства коллоидных растворов
В 1909 г профессор Московского университета Р. Рейсе наблюдал воздействие постоянного электрического тока на диспергированную в воде глину, и на этом основании описал электрические свойства коллоидных растворов. Частицы дисперсной фазы (глины) перемещались к аноду, где наблюдалось помутнение раствора, а частицы дисперсионной среды (воды) перемещались к катоду, где наблюдалось повышение уровня прозрачной жидкости. Направленное движение частиц к электродам говорило об их заряде. дисперсная фаза несет на себе заряд, противоположный по знаку заряду среды. Движение частиц дисперсной фазы к одному из электродов при пропускании через золь постоянного электрического тока получило название электрофореза, а движение частиц дисперсионной среды – электроосмоса. На границе раздела фаз возникает двойной электрический слой. Между фазами возникает разность потенциалов, называемая электродинамическим потенциалом Ψ. Часть скачка потенциала, обусловленная диффузным слоем, называется электрокинетическим или дзета – потенциалом ξ.
Дзета-потенциал определяется толщиной и зарядом диффузионного слоя, которые зависят от концентрации и заряда противоионов и температуры.
Чем меньше толщина диффузионного слоя, тем больше противоионов этого слоя перейдет за границу скольжения и тем меньше будет значение дзета-потенциала. Чем больше значение ξ, тем больше агрегатная устойчивость коллоидных систем.
Электрические свойства коллоидных растворов объясняют их агрегатную устойчивость, которая проявляется в том, что частицы дисперсной фазы в коллоидном растворе не укрупняются и не слипаются.
Коагуляция коллоидных растворов
Коагуляцией называют процесс соединения коллоидных частиц в крупные агрегаты с последующей потерей коллоидной системой кинетической устойчивости.
Коагуляцию коллоидных растворов можно вызвать нагреванием, охлаждением, интенсивным перемешиванием, а также добавлением различных электролитов. Добавление к коллоидному раствору электролитов приводит к снижению электрокинетического потенциала. Этот процесс характеризуется следующими закономерностями:
· Минимальная концентрация электролита, вызывающая коагуляцию коллоидного раствора, называется порогом коагуляции;
· Коагулирующим действием обладает не весь электролит в целом, а только тот его ион, который имеет заряд, одноименный с зарядом противоионов мицеллы;
· Коагулирующая способность иона зависит от его заряда. Ионы с большим зарядом (например, Fe3+) вызывают коагуляцию при гораздо меньших концентрациях, чем ионы с более низким зарядом (Fe2+);
· Коагулирующая способность ионов с одинаковым зарядом возрастает с увеличением радиуса иона. Ионы органических соединений всегда обладают более высокой коагулирующей способностью, чем неорганических;
· При увеличении концентрации электролита в растворе уменьшается электрокинетический потенциал, и коагуляция наступает при его определенном значении – критическом потенциале.
Коллоидные растворы в природе и технике
В природной воде содержится часть примесей в коллоидном состоянии. Поэтому воду, используемую для коммунальных нужд, электростанций, строительства, подвергают обработке, вызывающей коагуляцию коллоидных частиц.
Коагуляцию широко используют при очистке воды для удаления взвешенных веществ. В качестве коагулянтов используют сульфаты алюминия или железа.
Дымовые газы электростанций, металлургических заводов представляют собой аэрозоли. Для их коагуляции применяется электрогазоочистка методом электрофореза при очень высоких напряжениях. В коллоидном состоянии находятся многие составные части живых организмов: кровь, лимфа, внутриклеточная жидкость.
Наибольшее практическое применение имеют твердые системы с газовой дисперсной фазой, называемые твердыми пенами. На ж/д транспорте применяются такие газонаполненные пластмассы, как минора – для теплоизоляции изотермических, пассажирских и рефрижераторных вагонов, поролон – для изготовления мягких сидений в пассажирских вагонах, стипор - тепло- и звукоизоляционный материал в рефрижераторных вагонах, трубопроводах, стеновых панелях.
Тема: «Окислительно-восстановительные процессы»
План
1. Общие понятия и положения. Степень окисления
2. Процессы окисления и восстановления
3. Составление уравнений, метод электронного баланса
4. Типы окислительно-восстановительных реакций (ОВР)
5. Направление протекания ОВР
Все ионные реакции в растворах можно разделить на два типа. К первому из них относятся ионообменные реакции, протекающие без изменения заряда ионов, входящих в состав реагирующих веществ. Например, 2NaOH+H2SO4 = Na2SO4 + 2H2O. Ко второму типу относят реакции, идущие с передачей электронов от одних атомов другим – окислительно-восстановительные реакции (ОВР): например, 2KJ+Cl2 = 2KCl+J2.
К окислительно-восстановительным можно отнести реакции с раздельным протеканием окисления и восстановления (электрохимические).
Для характеристики состояния элементов в соединениях введено понятие степени окисления (СО). Под степенью окисления понимают воображаемый (формальный) заряд в соединении, вычисленный, исходя из предположения, что в соединении имеются только ионные связи. СО для большинства соединений имеет условный характер, так как не отражает реальный заряд атома, но, однако, широко используется в химии. В зависимости от электроотрицательности (ЭО) атома СО может быть отрицательной или положительной. Элементы с высоким значением ЭО имеют отрицательную СО, а с малым – положительную.
Определяют СО, исходя из следующих правил:
1. Степень окисления атомов в простом веществе равна нулю. Например, О20, О30, S80 и т.д.
2. В соединениях с ковалентными полярными связями более электроотрицательный элемент имеет следующие СО: F-, О-2 (за исключением пероксидов (Н2О2-), надпероксидов (КО2-1/2), озонидов (КО3-1/3) и O+2F2.
3. Элементы с малой ЭО проявляют СО: Н+ (за исключением солеобразных гидридов LiH-), щелочные металлы +1, щелочноземельные металлы +2.
4. Алгебраическая сумма СО элементов в нейтральной молекуле равна нулю, а в сложном ионе – заряду иона.
Большинство элементов в соединениях проявляют переменную СО.
Примеры. Fe+2O-2, Fe2+3O3-2, K+N+3O2-2, H+N+5O3-2.
Максимальная СО имеет периодическую зависимость от порядкового номера в периодической таблице элементов и совпадает с номером группы. Для неметаллов периодическую зависимость имеет также и минимальная СО – это определяется электронным строением атома.
Окисление – это процесс отдачи электронов атомом. Степень окисления при этом повышается: Zn – 2e = Zn2+.
Вещество, отдающее свои электроны, называется восстановителем (Zn). Zn2+ является окисленной формой цинка.
К типичным восстановителям относят простые вещества, атомы которых имеют низкое значение ЭО, например, металлы (особенно активные – щелочные и щелочноземельные), водород, углерод, анионы, атомы которых находятся в низшей СО, например, Cl-, NO2- и т.д.
Восстановление – это смещение электронов к веществу. СО при этом понижается. Например, Сu2+ + 2e = Cu. Вещество, принимающее электроны (Сu2+), называется окислителем. В данной реакции Cu является восстановленной формой, а Сu2+ - окисленной. К типичным восстановителям относят простые вещества, атомы которых имеют высокую ЭО (кислород, хлор, фтор), соединения кислорода – пероксиды (Н2О2), катионы и анионы, содержащие атомы с высокой СО (Fe3+, Pb4+, NO-3, CrO42-, ClO4-).
В химических реакциях окисление и восстановление – сопряженные процессы. В ходе ОВР восстановитель отдает свои электроны окислителю, сам при этом окисляется. Окислитель, принимая электроны, восстанавливается. Раздельное протекание окисления и восстановления происходит только в электрохимических процессах на электродах.
Метод электронного баланса. Уравнения ОВР часто имеют сложный характер, их составление, в частности, расстановка стехиометрических коэффициентов, представляет собой при этом трудную задачу. Одним из методов решения этой задачи является метод электронного баланса.
Рассмотрим этот метод на примере восстановления перманганата калия концентрированной соляной кислотой. Эта реакция используется в качестве лабораторного метода получения хлора.
Записываем схему реакции: KMnO4 + HCl = Cl2 + KCl + MnCl2 + H2O.
Определяем СО элементов: K+Mn+7O4-2 + H+Cl- = Cl20 + K+Cl- + MnCl2+ H2+O-2. В ходе взаимодействия только атомы марганца и хлора изменяют степень окисления. При этом степень окисления марганца уменьшается (Mn+7 – окислитель), а хлора – увеличивается (Cl- - восстановитель).
Составим схемы, отражающие процесс передачи электронов:
 Mn+7 + 5е = Mn+2 5 2
Mn+7 + 5е = Mn+2 5 2
2Cl- -2e= Cl20 2 5
Так как количества отданных и принятых электронов должно быть одинаковым, введем дополнительные множители, устанавливающие электронный баланс. Эти множители подберем по правилу нахождения наименьшего общего кратного, и они будут представлять собой коэффициенты при окислителе и восстановителе:
2KMnO4 + 10HCl = 5Cl2 + KCl + 2MnCl2 + H2O.
Далее уравняем количества атомов, не участвующих в процессах окисления-восстановления, в следующем порядке: количество атомов металлов (катионов), не изменивших степени окисления (калий); ионы кислотных остатков, не изменивших степени окисления (хлорид – анионы); количество ионов водорода; проверка правильности расстановки коэффициентов осуществляется подсчетом общего количества атомов кислорода слева и справа:
2KMnO4 + 16HCl = 5Cl2 + 2KCl + 2MnCl2 + 8H2O.
Типы ОВР. Рассмотренное выше уравнение описывает межмолекулярную ОВР. Окислитель и восстановитель являются разными веществами (атомы разных элементах находятся в разных молекулах).
Если окислитель и восстановитель являются атомами разных элементов, но находятся внутри одной молекулы, то реакция называется внутримолекулярной ОВР: 2AgNO3 = 2Ag + 2NO2 + O2.
Ag+ + e = Ag восстанавливается, окислитель
N+5 + e = N+4 восстанавливается, окислитель
2O-2 – 4e = O2 окисляется, восстановитель
2Ag+ + 2N+5 + 2O-2 = 2Ag + 2N+4 + O2
В некоторых ОВР происходит окисление и восстановление атомов или ионов одного и того же элемента. Такие реакции называются реакциями самоокисления-самовосстановления:
3H2MnO4 = 2HMnO4 + MnO2 + 2H2O
Mn+6 – e = Mn+7 2 восстановитель, окисляется
Mn+6 + 2e = Mn+4 1 окислитель, восстанавливается
2Mn+6 + Mn+6 = 2Mn+7 + Mn+4
Окислителем и восстановителем в данной реакции выступают ионы 2Mn+6, она является реакцией диспропорционирования. Если же эта реакция протекает в обратном направлении, то она будет называться реакцией конпропорционирования.
Направление ОВР. Возможность предсказать направление ОВР дает второе начало термодинамики. Если энергия Гиббса ОВР ниже нуля, то реакция может протекать в прямом направлении. Если энергия Гиббса ОВР выше нуля, то реакция в прямом направлении самопроизвольно не пойдет, а возможна в обратном направлении.
ОВР имеют важную роль в природе и технике. В природных биологических системах происходят реакции фотосинтеза в растениях, дыхания и метаболизма у животных и человека, которые являются окислительно-восстановительными. Процессы горения топлива, протекающие в топках котельных, ТЭЦ, двигателях внутреннего сгорания и реактивных двигателях, являются важными ОВР в технике и повседневной жизни. ОВР происходят при коррозии металлов, пожарах, окислении азота при сжигании топлива. Продуктами таких процессов часто являются вредные вещества, наносящие ущерб окружающей среде.
С помощью ОВР получают большинство простых веществ, в основном металлы, множество органических и неорганических веществ, а также проводят анализ и очищают вещества, например, сточные воды, металлы, газовые выбросы предприятий и т.д. В электронной и вычислительной технике, радио- и электротехнике, других областях науки и промышленности используют методы нанесения металлических покрытий в защитных и декоративных целях. ОВР лежат в основе действия топливных элементов, используемых в том числе в военной и космической технике.
Итак, ОВР являются широко используемыми реакциями, в которых происходит перемещение электронов от одних частиц к другим. Для составления уравнений используют несколько методов, мы рассмотрели метод электронного баланса. Направление ОВР можно предсказать, если известно изменение ΔG реакции.
ЛЕКЦИЯ ПО ТЕМЕ: «Электродные потенциалы и электродвижущие силы»
План
1. Электрохимические процессы
2. Понятие об электродном потенциале металла
3. Стандартный водородный электрод
4. Гальванические элементы. ЭДС гальванических элементов
5. Концентрационный гальванический элемент
Электрохимические процессы
Процессы прямого превращения химической энергии в электрическую и обратно называются электрохимическими процессами. Электрохимические процессы можно разделить на две основные группы: 1) процессы превращения химической энергии в электрическую (процессы в гальванических и топливных элементах); 2) процессы превращения электрической энергии в химическую (электролиз).
Электрохимическая система состоит из двух электродов и ионного проводника между ними. Электроды замыкаются металлическими проводниками. Ионным проводником (проводником второго рода) служат растворы или расплавы электролитов, а также твердые электролиты. Электродами называются проводники, имеющие электронную проводимость (проводники первого рода), находящиеся в контакте с ионным проводником.
Понятие об электродном потенциале металла
При погружении металлической пластины в воду или раствор соли данного металла на границе металл – раствор возникает скачок потенциала. Появление такого скачка связано с особенностями строения кристаллической решетки металлов, в узлах которой находятся положительные ионы, заряд которых уравновешивается зарядом электронного газа. Положительно заряженные ионы в поверхностном слое металла под действием полярных молекул воды отрываются и в гидратированном состоянии переходят в раствор, который при этом заряжается положительно. В самом металле появляется избыток электронов, придающих ему отрицательный заряд. На границе металл – раствор образуется двойной электрический слой, а между металлом и окружающей его средой создается разность потенциалов, которая называется электродным потенциалом. Чем активнее металл, тем больше разность потенциалов между металлом и раствором, тем больше скачок потенциала.
- + +
Металл - + + раствор
- + +
- + +
Двойной электрический слой на границе раздела металл – раствор
В двойном электрическом слое все время происходит движение ионов, причем одни из них входят обратно в кристаллическую решетку металла, другие снова переходят в раствор. Между раствором и поверхностью металла существует равновесие:
М+е– + nН2О ↔ М+ nН2О (в растворе) + е– (в металле).
Однако такое равновесие может быть смещено в ту или иную сторону.
Величина электродного потенциала зависит от ряда факторов, из которых основными являются природа металла (чем активнее металл, тем выше Е), природа растворителя (чем полярнее растворитель, тем больше Е) и концентрация ионов металла (повышение концентрации понижает Е).
Зависимость величины электродного потенциала от концентрации ионов металла и температуры выражается формулой Нернста:
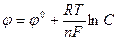
Здесь φ0 – нормальный (стандартный) электродный потенциал, возникающий на границе металл – раствор в том случае, когда металл помещен в раствор своей соли с концентрацией 1 моль/л, С – концентрация ионов металла, моль/л, R – универсальная газовая постоянная (8,314 Дж/моль˙град), Т – абсолютная температура, К, n – валентность иона, F – число Фарадея (96500 Кл/моль).
Подставляя в формулу Нернста величины R, F, значение Т = 298 К и переходя от натуральных логарифмов к десятичным, получаем сокращенную формулу Нернста: 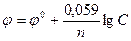 .
.
Она дает возможность рассчитать величину электродного потенциала при любой концентрации ионов металла в растворе при комнатной температуре.
Стандартный водородный электрод
В настоящее время за нулевое значение принят потенциал стандартного водородного электрода. Такой электрод состоит из платиновой пластины, контактирующей с газообразным водородом, находящимся под давлением 101 кПа, и раствором, в котором активность ионов водорода равна единице. Водородный электрод относится к газовым электродам. В качестве проводника первого рода для стандартного водородного электрода служит платина. При контакте платины с водородом происходит адсорбция водорода на платине. Адсорбированный водород, взаимодействуя с молекулами воды, переходит в раствор в виде ионов, оставляя в платине электроны. При платина заряжается отрицательно, а раствор – положительно. Возникает скачок потенциала между платиной и раствором. Наряду с переходом ионов в раствор идет обратный процесс восстановления ионов водорода с образованием молекул водорода.
Равновесие на водородном электроде можно представить в виде
2Н+ + 2е ↔ Н2
Абсолютное значение потенциала водородного электрода неизвестно, но условно считают за нуль потенциал стандартного водородного электрода, то есть потенциал при Р(Н2) = 1 атм (101 кПа) и аН+ = 1 моль/л.
Кроме водородного электрода, применяются и другие электроды сравнения.
Уравнение для расчета потенциала водородного электрода можно вывести также, как выведено уравнение для расчета потенциала металлического электрода. Для 298 К оно имеет вид
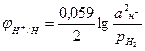
Где рН2 – парциальное давление водорода, аН+ - активность ионов водорода в электролите. Учитывая, что lgаН+=-рН, получим:
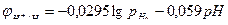
Потенциал водородного электрода принимает более отрицательное значение с увеличением давления водорода и рН.
Гальванические элементы. ЭДС гальванического элемента
Гальванический элемент состоит из двух электродов, в которых энергия химической реакции преобразуется в электрическую.
Примером может служить гальванический элемент Якоби-Даниэля. Он состоит из медной пластины, погруженной в раствор CuSO4, и цинковой пластины, погруженной в раствор ZnSO4. Для предотвращения прямого взаимодействия окислителя и восстановителя электроды отделены друг от друга пористой перегородкой. На поверхности цинковой пластины возникает двойной электрический слой и устанавливается равновесие: Zn–2e↔Zn2+. В результате протекания этого процесса возникает электродный потенциал цинка. На поверхности медной пластины также возникает двойной электрический слой и устанавливается равновесие: Cu–2e↔Cu2+. В результате возникает электродный потенциал меди.
Потенциал цинкового электрода имеет более отрицательное значение (φZn2+/Zn = -0,76 В) по сравнению с потенциалом медного электрода (φCu2+/Cu = 0,34 В). Цинковый электрод выполняет функцию анода, а медный – катода.
Итак, при работе элемента Якоби-Даниэля протекают следующие процессы:
1) Реакция окисления цинка на аноде: Zn–2e↔Zn2+.
2) Реакция восстановления меди на катоде: Cu2++2e↔ Cu.
3) Движение электронов во внешней цепи;
4) Движение ионов в растворе: анионов SO42- к аноду, катионов (Zn2+, Cu2+) к катоду.
Движение ионов в растворе замыкает электрическую цепь гальванического элемента. Суммируя электродные реакции, получим:
Cu2++ Zn↔ Cu+ Zn2+
Вследствие этой химической реакции в гальваническом элементе возникает движение электронов во внешней цепи и ионов внутри элемента, то есть электрический ток, поэтому суммарная химическая реакция в гальваническом элементе называется токообразующей реакцией.
Схема работы гальванического элемента:
Анод (–) Zn / Zn2+ // Cu2+ / Cu (+) Катод
Границу раздела между проводником первого рода и проводником второго рода обозначают одной чертой, а границу раздела между проводниками второго рода – двумя чертами.
Максимальная разность потенциалов электродов называется электродвижущей силой (ЭДС) элемента. Она равна разности равновесных потенциалов катода и анода элемента: ЭДС=φк – φа.
Например, для вычисления ЭДС медно-цинкового гальванического элемента надо от нормального электродного потенциала меди вычесть нормальный электродный потенциал цинка: ЭДС= φCu2+/Cu – φZn2+/Zn = 0,34-(-0,76) = 1,1 В. Чем больше отличаются металлы по величине своего электродного потенциала, то есть чем больше они отстоят друг от друга в ряду напряжений, тем больше ЭДС гальванического элемента, построенного из этих металлов. Движущей силой электрохимических процессов, протекающих в гальванических элементах, является изобарный потенциал (энергия Гиббса).
Связь между его изменением ΔG и ЭДС гальванического элемента Е выражают уравнением: ΔG= –nFE. Здесь n – число электронов, принимающих участие в окислительно-восстановительном процессе, F – число Фарадея. Чем больше ЭДС, тем больше отличается от нуля изменение изобарного потенциала ΔG.
Изменение потенциалов электродов при работе гальванических элементов называется гальванической поляризацией. Для её уменьшения применяют вещества, увеличивающие скорость катодных процессов. Такие вещества называют деполяризаторами. К ним относятся окислители (MnO2, O2, K2Cr2O7, Cu2+) и другие. Так, в медно-цинковом элементе деполяризатором служит раствор сульфата меди. Уменьшение поляризации гальванического элемента называют деполяризацией.
Концентрационные элементы
Гальванический элемент, в котором источником энергии является не химическая реакция, а работа выравнивания концентраций (активностей) ионов, называется концентрационным гальваническим элементом. Он состоит из двух одинаковых электродов, погруженных в растворы собственных солей разной концентрации, например: Cu / Cu2+ (а1) // Cu2+ (а2) / Cu, где а1<а2. Электрод, находящийся в более разбавленном растворе, является анодом. Он растворяется и его ионы переходят в раствор. На электроде, погруженном в более концентрированный раствор, осаждаются ионы металла – он является катодом. Таким образом, на обоих электродах идут процессы, приводящие к выравниванию концентраций ионов в растворах. Концентрационный элемент будет работать до тех пор, пока активности ионов в растворах у анода и катода не будут равны.
ЛЕКЦИЯ ПО ТЕМЕ: «Электролиз как окислительно-восстановительный процесс»
План
1. Сущность процесса электролиза
2. Электролиз водных растворов
3. Электролиз расплавов
4. Перенапряжение и поляризация
5. Законы Фарадея. Выход по току
6. Применение электролиза
Электролизом называют окислительно-восстановительный процесс, протекающий на электродах под действием постоянного электрического тока, подаваемого от внешнего источника, проходящего через раствор или расплав электролита. При электролизе происходит превращение электрической энергии в химическую.
Процесс электролиза производится в электролизере, куда заливают электролит и устанавливают два электрода. Электрод, на котором происходит реакция восстановления, называется катодом, подключается к отрицательному полюсу внешнего источника тока. Электрод, на котором происходит реакция окисления, называется анодом, подключается к положительному полюсу внешнего источника тока.
Схема электролиза
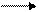 е- е-
е- е-

 анод + – катод
анод + – катод

Электролиз водных растворов
Характер химических реакций в водных растворах на катоде определяется положением металла в ряду стандартных электродных потенциалов. Чем меньше значение электродного потенциала металла, тем труднее восстанавливаются его ионы на катоде. По этому признаку их разделяют на три группы.
К первой группе относятся катионы металлов, находящиеся в ряду напряжений левее алюминия. Они не восстанавливаются на катоде из водных растворов, вместо них происходит восстановление ионов водорода (молекул воды): 2Н++2е=Н2.
Ко второй группе относятся катионы металлов, расположенных в ряду напряжений между алюминием и водородом. Они восстанавливаются одновременно с водородом: Zn2++2е= Zn; 2Н++2е=Н2.
К третьей группе относятся катионы металлов, находящиеся в ряду напряжений после водорода. Они восстанавливаются на катоде: Cu2++2е= Cu.
Окисление на аноде имеет свои закономерности. Анионы бескислородных кислот и их солей (Cl-, Br-, J-, S2-, CN- и т.п.) удерживают свои электроны слабее молекул воды, поэтому при электролизе окисляются в первую очередь. В то же время анионы кислородсодержащих кислот (NO3-, SO42-, PO43- и т.п.) удерживают свои электроны прочнее, чем молекулы воды, поэтому на аноде происходит окисление гидроксил-анионов: 4ОН- – 4е = 2Н2О + О2.
Характер реакций, протекающих на аноде, зависит также от природы вещества анода. Различают растворимые и нерастворимые аноды. нерастворимые аноды изготавливают из углерода (графита), платины, а растворимые – из меди, цинка, никеля, железа, свинца и их сплавов. Нерастворимый анод сам не претерпевает изменений, а лишь является передатчиком электронов. При применении растворимого анода происходит не окисление анионов из раствора, а растворение металла, то есть переход ионов металла, из которого изготовлен анод, в раствор. Этот метод используется для получения чистых металлов (электрорафинирование), а также для получения покрытий (гальваностегия).
Рассмотрим несколько примеров электролиза с нерастворимым анодом водных растворов солей.
1. Раствор сульфата натрия Na2SO4, a(Na+)=1M, a(SO42-)=1M. В растворе имеются ионы Na+, SO42-, Н+, ОН-.
Na2SO4 ↔ 2 Na+ + SO42- ; Н2О ↔ Н+ + ОН-.
Так как φ0(Na+/ Na)=-2,7В<φ0(Н+/ Н2), то на катоде будет восстанавливаться водород, а на аноде – кислород: в растворе 2Н2О ↔ 2Н+ + 2ОН-. На катоде 2Н++2е=Н2. На аноде 2ОН- – 2е = Н2О + 1/2 О2. Суммарной является реакция разложения воды: Н2О=Н2+1/2О2.
2. Раствор хлорида натрия NaCl, a(Na+)=1M, a(Cl-)=1M. В растворе имеются ионы Na+, Cl-, Н+, ОН-. NaСl ↔ Na+ + Cl-; Н2О ↔ Н+ + ОН-.
На катоде пойдет восстановление водорода: 2Н++2е=Н2. На аноде идет окисление ионов хлора: 2Cl- - 2е = Cl2.
Электролиз расплавов
Рассмотрим электролиз расплавов на примере электролиза расплава хлорида натрия. В расплаве хлорида натрия имеются ионы натрия и хлора: NaСl ↔ Na+ + Cl-; если погрузить в расплавленную соль два графитовых электрода и подключить их к полюсам внешнего источника тока, то в электролите начнется направленное движение ионов и на электродах будут протекать следующие реакции:
1) Катодный процесс – восстановление ионов натрия до металлического натрия (на отрицательном электроде) Na+ + е = Na+.
2) Анодный процесс – окисление ионов хлора на положительном электроде (с которого электроны идут во внешнюю цепь) 2Cl- - 2е = Cl2. Суммарная реакция 2NaCl = 2Na + Cl2.
Перенапряжение и поляризация
В процессе электролиза происходит изменение состава электродов с поверхности из-за образования на них продуктов электролиза. При этом образуется гальванический элемент, сила тока которого направлена обратно течению процесса электролиза. Это явление называется химической поляризацией. Для протекания электролиза необходимо приложить к электродам напряжение, большее, чем ЭДС образованного гальванического элемента. Наименьшая разность потенциалов, при которой протекает электролиз, называется потенциалом разложения Еразл.. Разность между потенциалом разложения и ЭДС гальванического элемента называется перенапряжением η.
Перенапряжение – это величина, которая показывает, насколько смещается электродный потенциал от равновесного значения.
Как катодное, так и анодное перенапряжение зависят от материала электродов, состояния их поверхности, от природы разряжающихся ионов и плотности тока.
Перенапряжение при электролизе может достигать значительной величины, что приводит к добавочному расходу электроэнергии. Поэтому стремятся создать такие условия, при которых величина η становится по возможности минимальной. Это достигается путем создания невысокой плотности тока.
Законы Фарадея
Электрохимические процессы на электродах подчиняются законам М. Фарадея.
1 закон Фарадея: массы веществ m, выделившихся на электродах при электролизе, прямо пропорциональны количеству электричества q, прошедшему через электролит: m=kq. Масса вещества, выделяющегося при прохождении одного кулона, называется электрохимическим эквивалентом k.
2 закон Фарадея: одинаковые количества электричества выделяют при электролизе на электродах эквивалентные массы веществ. Для выделения на электроде одного грамм-эквивалента любого вещества необходимо затратить одно и то же количества электричества, а именно 96500 Кл (называемого числом Фарадея).
Масса металла при электролизе на катоде, согласно законам Фарадея, может быть вычислена по формуле:
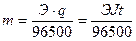 .
.
Здесь m – масса металла, г; Э – химический эквивалент металла, г/моль; t – продолжительность электролиза, с.
При электролизе во многих случаях выделяется веществ меньше, чем должно получиться по законам Фарадея. Это объясняется тем, что наряду с основными электродными процессами окисления и восстановления протекают побочные, параллельные процессы (в результате химической поляризации). Поэтому вводится понятие выхода по току χ, %. Оно представляет собой отношение массы полученного вещества в данных условиях электролиза mпр к массе, теоретически вычисленной на основании закона Фарадея mтеор:
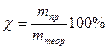
Применение электролиза
Электролиз широко используется в различных областях промышленности. Практически нет ни одной области техники, где бы он не применялся.
Электролиз в металлургии. Электролизом растворов солей получают медь, цинк, кадмий, никель, кобальт, марганец и другие металлы. На катоде происходит разряд ионов металла из раствора Mn+ + ne = M. В этих процессах используют нерастворимые аноды, на которых выделяется кислород: 2Н2О + 4е = О2 + 4Н+.
Метод электролиза используют для рафинирования металлов – меди, серебра, золота, свинца, олова и других. Анодом при рафинировании служит очищаемый металл. На аноде происходит растворение основного металла и примесей, потенциал которых отрицательнее потенциала основного металла. На катоде выделяется металл, имеющий более положительный потенциал. Так как потенциалы меди, серебра, свинца и олова положительнее, чем потенциалы других основных металлов ,то каждый из этих металлов в первую очередь выделяется на катоде, а примеси остаются в растворе.
Электролиз в химической промышленности. К наиболее крупномасштабному электролитическому процессу в химической промышленности относится электролиз раствора хлорида натрия с получением газообразных водорода и хлора на электродах и щелочи в электролизере. Кроме этого, электролизом получают фтор из расплава смеси плавиковой кислоты и фторида натрия, водород и кислород из воды, диоксид марганца из раствора сульфата марганца.
Получение гальванопокрытий. Гальваническими называются металлические покрытия, наносимые на поверхность какого-либо изделия методом электролиза. Гальваническим способом можно получить покрытия для всех металлов и сплавов. Наиболее распространены никелирование, хромирование, меднение и цинкование.
Нанесение гальванических покрытий проводится в электролизере, называемом гальванической ванной. Катодом служит изделие, на которое наносится покрытие. На катоде идёт процесс восстановления находящихся в растворе электролита ионов металла Mn+ + ne = M. Анодом обычно служит тот же металл, что и металл-покрытие. Процесс на аноде противоположен процессу на катоде: M – ne = Mn+
Электрохимическая анодная обработка металлов и сплавов. Анодная обработка изделий для придания им требуемой формы получила название электрохимической обработки металлов (ЭХОМ). Этот способ обработки металлов имеет важные достоинства, так как позволяет обрабатывать детали сложной конфигурации. Как и при обычном электролизе с растворимыми анодами, при ЭХОМ происходит анодное растворение металла M – ne = Mn+. На катоде, который называют инструментом, обычно выделяется водород 2Н++2е=Н2. Особенностью ЭХОМ по сравнению с другими методами электролиза является высокая скорость растворения металлов. В настоящее время ЭХОМ используется для обработки лопаток турбин, штампов и пресс-форм, твёрдых и тугоплавких металлов.
ЛЕКЦИЯ ПО ТЕМЕ: «Коррозия металлов»
План
1. Определение коррозии и причины её возникновения
2. Классификация коррозионных процессов. Химическая и электрохимическая коррозия
3. Коррозия металлов в различных средах
3.1. Контактная коррозия
3.2. Атмосферная коррозия
3.3. Подземная коррозия
3.4. Коррозия под действием блуждающих токов
4. Виды коррозионных разрушений
Определение коррозии и причины её возникновения
Коррозия – это самопроизвольный процесс разрушения металлов и сплавов в условиях природной среды.
При коррозии металлы окисляются и образуются продукты, состав которых зависит от условий среды.
Согласно современным представлениям, все основные изменения в органическом и неорганическом мире связаны с окислительно-восстановительными процессами. Окислительно-восстановительные реакции лежат в основе и коррозионных процессов.
Основной причиной коррозии является термодинамическая неустойчивость металлов и сплавов в окружающей среде. Подавляющее большинство металлов в земной коре находится в виде оксидов, сульфидов и других соединений. При получении металлов в металлургии их переводят из такого стабильного состояния в элементарную форму, которая нестабильна. При контакте металла с внешней окислительной средой появляется движущая сила, стремящаяся превратить их в стабильные соединения, подобные тем, которые находятся в рудах. Примером этого является коррозия стали. В результате этого элементарное железо превращается в окисленное двух- и трёхвалентное, которое соответствует таким минералам, как магнетит (Fe3O4) или лимонит (Fe2O3˙H2O).
Термодинамическая неустойчивость металлов количественно оценивается знаком и величиной изобарно-изотермического потенциала ΔG (энергии Гиббса). Самопроизвольно протекают те процессы, которые сопровождаются уменьшением энергии Гиббса, то есть для которых ΔG<0. Металлы, стоящие в ряду напряжений до водорода, имеют по сравнению с водородом более отрицательный потенциал, их окисленное состояние термодинамически более устойчиво, чем восстановленное. Для металлов, расположенных после водорода, восстановленное состояние термодинамически более устойчиво, то есть для них ΔG>0. К этой группе металлов относятся коррозионно-стойкие золото, платина, серебро и др.
Классификация коррозионных процессов. Химическая и электрохимическая коррозия
Коррозионные процессы классифицируются:
1. По механизму реакций взаимодействия металла со средой;
2. По типу коррозионной среды;
3. По характеру коррозионных разрушений на поверхности и в объёме металла;
4. По характеру механических воздействий, которым подвергается металл одновременно с действием коррозионной среды.
По первому признаку различают два вида коррозии – химическую и электрохимическую.
Химическая коррозия
Химическая коррозия протекает при взаимодействии металлов с окислителями в средах, не проводящих электрический ток. Механизм химической коррозии можно представить одностадийным процессом окисления металла, то есть взаимодействием поверхности металла с окислителем.
Химическая коррозия – это процесс самопроизвольного разрушения металла в среде окислительного газа (например, кислорода) при повышенной температуре. Скорость химической коррозии зависит от многих факторов, в первую очередь она определяется характером продуктов коррозии. При окислении на поверхности металла образуется твердая пленка оксидов. Скорость окисления определяется состоянием и защитными свойствами поверхностной плёнки. Это зависит от отношения объёмов оксидной плёнки Vок и прокорродированного металла Vм, из которого она образовалась. Установлено, что для пористых плёнок, не предохраняющих металл от доступа агрессивных примесей воздуха 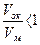 . А для плёнок, обладающими защитными свойствами,
. А для плёнок, обладающими защитными свойствами, 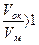 .
.
Скорость химической коррозии возрастает с увеличением температуры из-за повышения коэффициента диффузии и изменения защитных свойств плёнки. Резкие изменения температуры часто вызывают быстрое разрушение защитной плёнки. Это связано с различными коэффициентами термического расширения металла и плёнки.
По условиям протекания коррозионного процесса различают газовую коррозию (протекающую в газах, парах при высокой температуре в отсутствие воды), и коррозию в жидкостях – неэлектролитах (нефть, фенол, бензин, бензол).
Электрохимическая коррозия
При электрохимической коррозии процесс взаимодействия металла с окислителем состоит из двух сопряженных реакций: анодного растворения металла и катодного восстановления окислителя. Эта коррозия может протекать в электролитах, атмосфере любого влажного газа, а также в почве.
Основным отличием электрохимической коррозии от химической является наличие влаги на поверхности металла, что приводит к контакту двух различных металлов через электролит. При этом возникают короткозамкнутые гальванопары, в результате чего появляется электрический ток. В этом случае процесс коррозии обусловлен работой гальванопары, то есть электрохимической реакцией. По этой причине электрохимическая коррозия более агрессивна по отношению к металлам, чем химическая.
Механизм электрохимической коррозии состоит в том, что происходит анодное окисление металла: M – ne = Mn+ и катодное восстановление окислителя (Ох) Ох+ne=Red.
Окислителями при коррозии служат молекулы кислорода, хлора, ионы Н+, Fe3+, NO3– и др. Наиболее часто при коррозии наблюдается ионизация (восстановление) кислорода в нейтральной (щелочной) среде О2+2Н2О+4е=4ОН–, в кислой среде – восстановление водорода 2Н++2е=Н2.
Коррозия с участием кислорода называется коррозией с поглощением кислорода или коррозией с кислородной деполяризацией. Коррозия с участием ионов водорода называется коррозией с выделением водорода или коррозией с водородной деполяризацией.
Кроме первичных реакций, в растворе протекают вторичные реакции:
Мх++хОН-=М(ОН)х
В результате взаимодействия металла с кислородом, как и при химической коррозии, образуется оксид металла: М(ОН)2=МО+Н2О.
Кроме анодных и катодных реакций при электрохимической коррозии происходит движение электронов в металле и ионов в электролите. Электролитами могут быть растворы солей, кислот и оснований, морская и атмосферная вода (содержащая кислород, углекислый, сернистый и др. газы). Основным отличием электрохимической коррозии от процессов в гальваническом элементе является отсутствие внешней цепи.
Равновесные потенциалы водородного и кислородного электродов в зависимости от рН среды находят на основании уравнения Нернста:
φ2Н+/Н2=-0,059рН;
φО2/ОН=1,23-0,059рН.
Коррозия металлов в различных средах
Контактная коррозия
Контактная биметаллическая коррозия является разновидностью электрохимической коррозии, вызванной контактом металлов, имеющих разные электродные потенциалы в электролите. При этом коррозия металла с более отрицательным потенциалом обычно усиливается, а разрушение металла с положительным потенциалом замедляется или полностью прекращается. При конструировании учитывают возможность контактов различных металлов.
Атмосферная коррозия
На скорость атмосферной коррозии влияет влажность и газовый состав атмосферы. Влажность ,температура и степень загрязнения атмосферы влияют на качество и состав образующихся на поверхности металла плёнок. Наиболее агрессивны среды, сильно загрязненные промышленными газами (СО2, SO2, NO2, NH3, HCl), частицами солей и угольной пылью. В промышленных районах атмосферную коррозию могут интенсифицировать так называемые «кислотные дожди», основными агрессивными компонентами которых являются серные и азотные кислоты. Кислотные дожди (рН<4) легко вызывают коррозию сплавов алюминия, железа и цинка.
В зависимости от влажности атмосферы различают несколько видов атмосферной коррозии: мокрую, влажную и сухую. Мокрая атмосферная коррозия при относительной влажности до 100% наблюдается при наличии адсорбционной капиллярной или химической плёнки влаги на поверхности металла. Её толщина составляет от 0,1 мм до 1 мм. Понижение температуры интенсифицирует процесс конденсации и приводит к появлению капель влаги на поверхности металла.
Влажная коррозия возникает при влажности в атмосфере ниже 100%. Толщина плёнки влаги от 100 А0 до 0,1 мм. При влажности воздуха менее 60% наблюдается сухая атмосферная коррозия (коррозия под действием кислорода воздуха). Процесс разрушения металла подчиняется законам, характерным для газовой коррозии.
Подземная коррозия
Коррозионные разрушения металлических конструкций в почвах и грунтах вызываются подземной коррозией. Ей подвержены трубопроводы (водные, газовые, нефтяные), опоры электроконтактной сети и др. Скорость коррозии зависит от пористости и состава почвы, величины рН, наличия микроорганизмов. Подземная коррозия протекает по механизму электрохимической коррозии. Почвенная влага играет роль электролита и процесс коррозии протекает следующими образом:
Анодная реакция Fe-2e=Fe2+
Катодная реакция О2+2Н2О+4е=4ОН–
Реакции в почве Fe2++2OH-=Fe(OH)2, 4Fe(OH)2+2H2O+O2=4Fe(OH)3, 2Fe(OH)3+(n-3)H2O=Fe2O3nH2O.
Поверхность металла в местах ограниченного доступа кислорода выполняет роль катода.
Грунтовая коррозия металлических конструкций чаще всего происходит в условиях, характерных для нейтральных сред, с участием кислорода в качестве деполяризатора. В кислых почвах может происходить коррозия с водородной деполяризацией.
Исследование коррозионной активности грунтов позволили сделать вывод о том, что наиболее коррозионно-активными являются болотистые почвы, торфяники, ил. Песок и известняк практически не коррозионно-активны. Существенное влияние на скорость коррозии металлов оказывает рН почвы. В почвах с рН меньше 6,5 коррозионная активность по отношению к стали повышается. Наибольшей коррозионной активностью обладают почвы с рН<5,5. Нейтральные почвы с рН=6,5–7,5 и слабощелочные до рН=8,5 не коррозионно-активны.
На скорость коррозии влияет также величина удельного электрического сопротивления грунта. Коррозия металлических подземных конструкций зависит от содержания в почве и грунте различных солей. Так, с увеличением содержания хлоридов, сульфатов скорость коррозии возрастает. Повышение температуры также способствует повышению скорости грунтовой коррозии металлов.
Коррозия под действием блуждающих токов
Блуждающими токами называются электрические токи, протекающие в земле при использовании её в качестве токопроводящей среды. Попадая в металлические конструкции, расположенные в грунте, они вызывают коррозию. Источниками возникновения блуждающих токов в почве являются электрифицированные железные дороги постоянного тока, трамваи, линии электропередач.
Поскольку рельсы не достаточно изолированы от земли, а почва является проводником, то часть тока уходит в землю, встречая на своём пути подземные металлические сооружения. Так как контактный провод подсоединен к положительному полюсу тяговой подстанции, а рельс – к отрицательному, то в месте выхода тока из рельса образуется анодная зона, где коррозия разрушает подошву рельса и крепежные детали. При этом, чем меньше переходное сопротивление рельс-земля, тем большая часть тока возвращается к тяговой подстанции через землю и тем интенсивнее анодная зона на рельсе. Этот вид коррозии очень опасен, так как блуждающие токи нередко распространяются на несколько десятков километров и вызывают сильные повреждения металлических конструкций.
Виды коррозионных разрушений
По виду коррозионного разрушения коррозия делится на следующие виды.
1. Сплошная, или общая коррозия. Она может быть равномерной, если фронт коррозионного разрушения распределяется параллельно плоскости металла, и неравномерной, когда скорость коррозии на различных участках неодинакова.
2. Избирательная коррозия. Она характерна для сплавов и твердых растворов.
3. Локальная коррозия. Она связана с образованием и локализацией пораженных коррозией мест в виде «раковин» разной величины.
4. Питтинг - коррозия. Разрушение металла начинается в глубине, с образованием пор; часто приводит к образованию сквозных отверстий.
5. Межкристаллитная коррозия. Разрушение идет по границам металлических кристаллов.
6. Внутрикристаллическая коррозия. Наблюдается при коррозионном растрескивании под действием внешних механических нагрузок или внутренних напряжений.
ЛЕКЦИЯ ПО ТЕМЕ: «Защита металлов от коррозии»
План
1. Легирование металлов
2. Защитные покрытия
3. Электрохимическая защита (катодная, анодная, протекторная). Защита от коррозии блуждающими токами
4. Ингибиторы коррозии
Все методы защиты металлов от коррозии условно делятся на следующие группы: легирование металлов, защитные покрытия, электрохимическая защита, изменение свойств коррозионной среды, рациональное конструирование изделий.
Легирование металлов
Это эффективный метод повышения коррозионной стойкости металлов. При легировании в состав сплава вводят компоненты, вызывающие пассивацию металла. В качестве таких компонентов применяют хром, никель, вольфрам и другие металлы. Широкое применение нашло легирование для защиты от газовой коррозии. Введение некоторых добавок в стали (титана, меди, хрома и никеля) приводит к тому, что при коррозии образуется плотная плёнка продуктов реакции, предохраняющая сплав от дальнейшей коррозии. При этом обеспечивается жаростойкость и жаропрочность сплавов.
Жаростойкость обычно обеспечивается легированием металлов и сплавов (например, стали хромом, алюминием и кремнием). Эти элементы при высоких температурах окисляются энергичнее, чем железо, и образуют плотные защитные плёнки оксидов, например, SiO2, Al2O3, Cr2O3. Хром и кремний также улучшают жаропрочность сталей. Легирование также используется с целью снижения скорости электрохимической коррозии, особенно коррозии с выделением водорода. К коррозионностойким сплавам относятся нержавеющие стали, в которых легирующими компонентами служат хром, никель и другие металлы.
Защитные покрытия
Слои, искусственно создаваемые на поверхности металлических изделий для предохранения их от коррозии, называются защитными покрытиями. Применяемые в технике покрытия подразделяются на металлические и неметаллические.
Металлические покрытия. Материалами для металлических защитных покрытий могут быть как чистые металлы (цинк, кадмий, алюминий, никель, медь, олово, хром, серебро), так и их сплавы (бронза, латунь и др.). По характеру поведения металлических покрытий при коррозии их можно разделить на анодные и катодные.
К катодным покрытиям относятся покрытия, потенциал которых в данной среде имеют большее значение, чем потенциал основного (покрываемого) металла. В качестве примеров катодных покрытий для стали можно привести медь, никель, кадмий, олово, серебро. При повреждении покрытия возникает коррозионный элемент, в котором основной материал (сталь) служит анодом и растворяется, а материал покрытия – катодом, на котором выделяется водород или поглощается кислород. Следовательно, катодные покрытия могут защищать металл от коррозии лишь при отсутствии пор и повреждений покрытия.
Анодные покрытия имеют меньший потенциал, чем потенциал основного металла. Примером анодного покрытия может служить цинк на стали. В этом случае основной металл будет катодом коррозионного элемента, поэтому он не корродирует.
Для получения металлических защитных покрытий применяются различные способы: электрохимический (гальванические покрытия), погружение в расплавленный металл, термодиффузионный и химический.
Неметаллические защитные покрытия. Они могут быть как неорганическими, так и органическими. Защитное действие этих покрытий сводится в основном к изоляции металла от окружающей среды. В качестве неорганических покрытий применяют неорганические эмали, оксиды металлов, соединения хрома, фосфора и др. К органическим относятся лакокрасочные покрытия, покрытия смолами, полимерными плёнками, резиной.
Электрохимическая защита
Электрохимическую защиту используют для предотвращения разрушения подземных трубопроводов, кабелей, корпусов судов, резервуаров, подводных лодок и т.д.
Электрохимическая защита основана на замедлении катодных и анодных реакций микрогальванических элементов. Она осуществляется присоединением к конструкции источника постоянного тока или дополнительного электрода.
Электрохимическую защиту подразделяют на катодную и анодную.
Катодная защита – наиболее распространенный вид электрохимической защиты. Её используют для борьбы с коррозией таких металлов и сплавов, как сталь, медь, латунь, алюминий в условиях не очень агрессивных сред. Она эффективна для предотвращения коррозионного растрескивания, обесцинкования латуней, питтинга сталей в почвах и морской воде. Наибольшее применение катодная защита получила для борьбы с коррозией подземных сооружений – трубопроводов, газопроводов, кабельных установок.
Катодную поляризацию можно осуществить путём присоединения защищаемой конструкции к отрицательному полюсу (катоду) внешнего источника тока или к металлу, имеющему меньший электродный потенциал. Положительный полюс подсоединяется к вспомогательному электроду, аноду. В процессе защиты анод активно разрушается и подлежит периодическому обновлению. В качестве материла анода применяют лом чугуна, стали, графита и т.п.
Защитный эффект можно оценить по формулам:
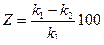 ,
, 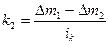 .
.
Здесь z – защитный эффект, к1 – показатель скорости коррозии металла без катодной защиты, к2 – при катодной защите, Δm1 – уменьшение массы металла без катодной защиты, Δm2 – при катодной защите, ik – катодная плотность тока.
Протекторная защита. К защищаемой конструкции присоединяют более электроотрицательный металл – протектор, который, растворяясь в окружающей среде, посылает электроны и катодно поляризует конструкцию. После полного растворения протектора или потери контакта его с защищаемой конструкцией протектор необходимо возобновлять. В качестве протектора чаще всего используют сплавы магния и цинка. Алюминий применяется реже, так как он быстро покрывается очень плотной оксидной плёнкой, которая пассивирует его и ограничивает токоотдачу. Протектор работает эффективно, если его переходное сопротивление (между ним и окружающей средой) невелико. В процессе работы протектор может покрываться слоем продуктов коррозии, которые изолируют его от окружающей среды и резко увеличивают переходное сопротивление. Для борьбы с этим протектор помещают в наполнитель (смесь солей), облегчающую растворение продуктов коррозии. Действие протектора ограничивается определенным расстоянием (радиусом действия). В настоящее время протекторную защиту применяют для борьбы с коррозией металлических конструкций в морской и речной воде, грунте и других нейтральных средах. Использование протекторной защиты в кислых средах ограничивается высокой скоростью саморастворения протектора.
Анодная защита. Скорость электрохимической коррозии металла может быть уменьшена и при его анодной поляризации, если она смещает потенциал защищаемого металла в пассивную область.
Метод анодной защиты имеет относительно ограниченное применение, так как пассивация эффективна в основном в окислительных средах при отсутствии активных ионов (например, ионов хлора для железа). Кроме того, анодная защита потенциально опасна: в случае прерывания подачи тока возможно активирование металла и его интенсивное анодное растворение. поэтому анодная защита требует тщательной системы контроля. Защитная плотность тока достаточно низкое и потребление электроэнергии невелико. Другое достоинство анодной защиты – высокая рассеивающая способность, то есть возможность защиты на более удаленном от катода расстоянии и в электрически экранированных участках.
Метод анодной защиты используют для металлов и сплавов, легко пассивирующихся при анодной поляризации; в химической промышленности – для снижения скорости коррозии низкоуглеродистой стали в серной кислоте и в растворах, содержащих аммиак и нитрат аммония.
Защита от коррозии блуждающими токами
Борьба с коррозией блуждающими токами заключается в их уменьшении. Это достигается:
1) Поддержанием в хорошем состоянии контактов между рельсами;
2) Увеличением сопротивления между рельсом и землей (использование шпал, применение щебёночного балласта);
3) Электродренажной защитой. Она обеспечивается путём отвода блуждающих токов от металлического сооружения в сторону их источника. Для этого подземное металлическое сооружение через дренажное устройство соединяется с отрицательной шиной или отсасывающей линией;
4) Применением токоотводов. С этой целью анодные зоны (например, на трубопроводе) с помощью медного проводника соединяют с чугунным ломом (анодом). В результате блуждающие токи вызывают коррозию только этого лома – анода.
Ингибиторы коррозии
Уменьшить коррозию металлической аппаратуры, например, в теплообменных трубках охлаждения дизелей на тепловозах, можно введением в агрессивную среду соединений, значительно снижающих коррозионный процесс. Такой способ снижения скорости коррозии называется ингибированием, а вводимые в среду вещества ингибиторами ил замедлителями коррозии.
Итак, ингибиторы – это такие вещества, введение небольших количеств которых в коррозионную среду, упаковочные средства и во временные защитные покрытия снижает скорость коррозии и уменьшает ее вредные последствия. Защитное действие ингибиторов связано с изменением в состоянии поверхности защищаемого металла и в кинетике реакций, лежащих в основе коррозионного процесса.
Скорость коррозии благодаря введению ингибитора может быть снижена в любое желаемое число раз, а степень защиты доведена почти до 100%. Эффективность ингибитора определяется как его природой, так и природой корродирующего металла, и зависит от температуры.
Ингибиторы коррозии можно классифицировать по различным признакам.
1. По составу их подразделяют на две группы: неорганические и органические. В последнее время широко применяют метало- и кремнийорганические ингибиторы.
2. По областям применения ингибиторы бывают: кислотной коррозии, щелочной коррозии и коррозии в нейтральных средах.
3. По условиям применения – существуют низкотемпературные и высокотемпературные ингибиторы.
4. По особенностям механизма действия ингибиторов бывают адсорбционные и ингибиторы пассивирующего действия.
Ингибиторы коррозии пассивирующего действия для нейтральных сред делятся на:
¨ Ингибиторы окислительного типа, которые проявляют своё действие и в отсутствие кислорода воздуха. Примеры: нитрит натрия NaNO2, нитрит аммония NH4NO2, хромат калия K2CrO4, дихромат калия K2Cr2O7, молибдат натрия Na2MoO4 и др.
¨ Ингибиторы, не обладающие окислительными свойствами, которые для проявления своего действия нуждаются в кислороде воздуха. Примеры: гидроксид аммония NH4NO3, гидроксид натрия NaOH, карбонат натрия Na2CO3, силикат, ортофосфат и тетраборат натрия Na2SiO3 , Na3PO4 и Na2B4O7.
Ингибиторы пассивирующего действия в нейтральных средах окислительного типа при отсутствии хлоридов и сульфатов по отношению к низкоуглеродистым сталям обладают примерно в сто раз большей эффективностью, чем ингибиторы, не обладающие окислительными свойствами. Наименьшая защитная концентрация ингибиторов–окислителей составляет 10–3 ¸ 10–4 %, а ингибиторов, не обладающих окислительными свойствами – 0,1 ¸ 0,05%. Ингибиторы коррозии могут вводиться в жидкие среды любой кислотности и в твёрдые материалы: масла, топлива, различные органические жидкости, лакокрасочные, полимерные, фосфатные, оксидные и другие покрытия, а также в упаковочные материалы. Наиболее перспективным является внесение в упаковочные материалы летучих ингибиторов (бензоат аммония, бензоат триэтаноламина, уротропин в смеси с нитритом натрия, нитрит дициклогексиламмония), которые, испаряясь в атмосферу внутри упаковки и адсорбируясь на поверхности металла, переводят его в пассивное состояние.
Замедление скорости коррозии связано прежде всего с исключением из коррозионного процесса части поверхности из-за её экранирования ингибитором. При выборе ингибиторов следует исходить не только из того, как они уменьшают скорость перехода металла в окружающую среду, но и из того, как они влияют на металлические свойства. Поверхностно-активные органические вещества (ПАОВ) с преобладающей катионной функцией более предпочтительны, чем с анионной функцией. Применение подобранных ингибиторов может не только предотвратить растворение металла, но и улучшить его механические свойства.
Защитное действие пассивирующих ингибиторов основано на смещении потенциала металла в положительную сторону и переводе его в пассивное состояние. Такой эффект может быть достигнут различными путями, но во всех случаях причиной снижения скорости коррозии является образование поверхностного защитного слоя. Ингибиторы могут непосредственно участвовать в образовании этого слоя.
Ингибиторы кислотной коррозии применяют при травлении изделий из черных и цветных металлов для удаления с их поверхности окалины и ржавчины, кислотной промывке теплосилового оборудования, при производстве кислот.
Действие ингибиторов атмосферной коррозии так же, как и других типов ингибиторов, сводится прежде всего к изменению ими кинетики электрохимических реакций, лежащих в основе коррозии. Эффективность любых ингибиторов зависит от их концентрации в коррозионной среде, и при некоторых минимальных значениях падает до нуля. Объем окружающей нас воздушной атмосферы практически безграничен и содержание в ней защитной концентрации ингибитора представляется экономически бессмысленно. применение ингибиторов для защиты металлов от атмосферной коррозии возможно поэтому лишь в том случае, если удается ограничить пространство, в которое помещается защищаемый объект, и отделить его от остальной атмосферы. Для этого ингибиторы вводят в смазки, полимерные и другие покрытия; помещают металл в упаковочный материал с внесением ингибитора в свободное пространство между упаковочным материалом и металлическим изделием, или в сам упаковочный материал (например, бумагу).
ЛЕКЦИЯ ПО ТЕМЕ: «Полимеры»
План
1. Методы получения полимеров
2. Строение полимеров
3. Свойства полимеров
4. Применение полимеров
Полимеры – высокомолекулярные соединения, которые характеризуются молекулярной массой от нескольких тысяч до нескольких миллионов. Молекулы полимеров (макромолекулы) состоят из большого числа повторяющихся звеньев. Отдельную группу соединений составляют олигомеры, которые по значению молекулярной массы занимают промежуточное положение между низкомолекулярными и высокомолекулярными соединениями. Различают неорганические, органические и элементоорганические полимеры. Органические полимеры в свою очередь подразделяют на природные и синтетические.
Методы получения полимеров
Полимеры получают методами полимеризации и поликонденсации.
Полимеризация – это реакция образования полимеров путём последовательного присоединения молекул низкомолекулярного вещества (мономера). При полимеризации не образуются побочные продукты и соответственно элементный состав макромолекул не отличается от состава молекул мономеров. В качестве мономеров используют соединения с кратными связями: С≡С, С≡N, C=C, C=O, C=N, либо соединения с циклическими группировками, способными раскрываться. В процессе полимеризации происходит разрыв кратных связей или раскрытие циклов у мономеров и возникновение химических связей между группами с образованием макромолекул, например:
n СН2=СН2 → (–СН2–СН2–)n ; n СН2=СН(С6Н5) → (–СН2–СН(С6Н5)–)n
Этилен Полиэтилен Стирол Полистирол
n RC≡N → (–C(R)=N–)n ; n CH2=CCH3COOCH3 → (–CH2–C(CH3)–)n
нитрил полинитрил COOCH3
метилметакрилат полиметилметакрилат
 n CH2–CH2 → (–CH2–CH2O–)n ; n CH≡CH → (–CH=CH–)n
n CH2–CH2 → (–CH2–CH2O–)n ; n CH≡CH → (–CH=CH–)n
O ацетилен полиацетилен
Этиленоксид полиэтиленоксид
n CH2=CHCH=CH2 → (–CH2–CH=CH–CH2–)n
бутадиен полибутадиен
По числу участвующих мономеров различают гомополимеризацию (один вид мономера) и сополимеризацию (два и более видов мономеров). Полимеризация – самопроизвольный экзотермический процесс (ΔG<0, ΔH<0), так как разрыв двойных связей или циклов с образованием одинарных связей ведет к уменьшению энергии системы. Однако без внешнего воздействия (инициаторов, катализаторов, нагревания или облучения) полимеризация если и протекает, то очень медленно. Полимеризация является цепной реакцией. В зависимости от характера активных частиц различают радикальную и ионную полимеризацию.
При радикальной полимеризации процесс инициируется свободными радикалами и проходит через несколько стадий: инициирование, рост цепи, передача или обрыв цепи. Радикальная полимеризация служит промышленным способом синтеза таких полимеров, как поливинилхлорид (-СН-СНСl-)n, поливинилацетат (-СН2-СН(ОСОСН3)-)n, полистирол (-СН2-СН(С6Н5)-)n, полиакрилат (-СН2-СН(СН3)(СООR)-)n, полиэтилен (-СН2-СН2-)n, полидиены (-СН2-СR=CH-CH2-)n и различных сополимеров.
Ионная полимеризация также проходит через стадию образования активных центров, роста и обрыва цепи. Роль активных центров играют катионы и анионы и соответственно различают катионную и анионную полимеризацию. Метод ионной полимеризации используют в производстве полиизобутилена (-СН2-С(СН3)2-)n, полиформальдегида (-СН2О-)n, полиамидов, например, поликапроамида (капрона) (-NH-(CH2)5-CO-)n, синтетических каучуков, например, бутадиенового каучука (-СН2-СН=СН-СН2-)n. Методом полимеризации получают ¾ всего объёма выпускаемых полимеров. Полимеризацию проводят в массе, растворе, эмульсии, суспензии или в газовой фазе.
Полимеризация в массе (блоке) – это полимеризация жидкого мономера в неразбавленном состоянии, при этом получают достаточно чистый мономер. Эмульсионная полимеризация (в эмульсии) происходит при полимеризации мономера, диспергированного в воде, в присутствии ПАВ для стабилизации эмульсии. Достоинство способа – лёгкость отвода теплоты, возможность получения полимеров с большой молекулярной массой, высокая скорость реакции; недостаток – необходимость отмывки полимера от эмульгатора. Способ применяется в промышленности для получения каучуков, полистирола, поливинилхлорида, поливинилацетата, полиметилметакрилата и других полимеров.
При полимеризации в суспензии полимер находится в виде капель, диспергированных в воде или другой жидкости. В результате реакции образуются полимерные гранулы размером от 10-6 до 10-3 м. При газовой полимеризации мономер находится в газовой фазе, а полимерные продукты – в жидком или твёрдом состоянии. Метод применяют для получения полипропилена и других полимеров.
Поликонденсация – это реакция синтеза полимера из соединений, имеющих две или более функциональные группы, сопровождающаяся образованием побочных низкомолекулярных продуктов (воды, аммиака, хлороводорода и др.). Поэтому элементные составы полимеров и исходных веществ не совпадают. Этим поликонденсация отличается от полимеризации. Поликонденсация протекает по ступенчатому механизму, при этом промежуточные продукты являются стабильными, то есть поликонденсация может остановиться на любой стадии. Образующиеся низкомолекулярные продукты реакции могут взаимодействовать с промежуточными продуктами поликонденсации, вызывая их расщепление (гидролиз, аминолиз, ацидолиз и т.д.).
Поликонденсацию проводят либо в расплаве, либо в растворе, либо на межфазной границе. Методом поликонденсации получают примерно ¼ часть выпускаемых полимеров, например, поликапроамид (капрон), полигексаметиленадипинамид (найлон) (-NH(CH2)6-NHCO(CH2)4CO-)n, полиэфиры - полиэтилентерефталат (-ОСС6Н4(СО)ОСН2СН2-)n, полиуретаны (-OROCONHR/NHCO-)n, полисилоксаны (-SiR2-O-)n, полиацетали (-OROCHR/-)n, фенолформальдегидные смолы (-C6H3OH-CH2-)n, мочевиноформальдегидные смолы и другие.
Строение полимеров
Форма и структура макромолекул
Макромолекулы могут быть линейными, разветвленными и сетчатыми. Линейные полимеры образуются при полимеризации мономеров или линейной поликонденсации. Разветвлённые полимеры могут образоваться как при полимеризации, так и при поликонденсации. Сетчатые полимеры образуются в результате сшивки цепей при вулканизации, образовании термореактивных смол и так далее. Форма макромолекул влияет на структуру и свойства полимеров.
Линейные и разветвлённые макромолекулы из-за способности атомов и групп атомов вращаться вокруг одинарных связей постоянно изменяют свою пространственную форму, или, другими словами, имеют много конформационных структур. Это свойство обеспечивает гибкость макромолекул, которые могут изгибаться, скручиваться, распрямляться. Поэтому для линейных и разветвленных полимеров характерно высокоэластичное состояние, то есть способность к обратимой деформации под действием относительно небольших внешних сил. Они также обладают термопластичными свойствами, то есть способны размягчаться при нагревании и затвердевать при охлаждении без химических превращений. При разветвлении полимеров эластические термопластичные свойства уменьшаются. При образовании сетчатой структуры термопластичность теряется.
Кристаллическое состояние полимеров.
Некоторые полимеры в определенных условиях могут иметь кристаллическую структуру. Способны кристаллизоваться только стереорегулярные полимеры. Процесс кристаллизации протекает через несколько стадий. 1 стадия – возникновение пачек – ассоциатов упорядоченно расположенных молекул. Из пачек образуются фибриллы и сферолиты. Фибриллы представляют собой агрегаты пачек продолговатой формы, а сферолиты – игольчатые образования, радиально расходящиеся из одного центра. Наконец, из фибрилл и сферолитов образуются единичные кристаллы. Между кристаллами находятся участки с неупорядоченной структурой (аморфные области). Поэтому такие полимеры характеризуются определенной степенью кристалличности, например, степень кристалличности полиэтилена может достигать 80%.
Физическое состояние аморфных полимеров
Аморфные полимеры находятся в стеклообразном, высокоэластичном и вязкотекучем состояниях. Для определения температурных границ существования этих состояний изучают зависимость деформации полимера от температуры, на основании которой строят термохимическую кривую.
 Деформация
Деформация
Термомеханическая кривая полимеров

I II III
 Тст Тт Тр температура
Тст Тт Тр температура
При низкой температуре полимер находится в стеклообразном состоянии (область I), в котором полимер ведёт себя как упругое твёрдое тело. При повышении температуры полимер переходит в высокоэластичное состояние, свойственное высокомолекулярным соединениям (область II). Высокоэластичное состояние проявляется в интервале температур от Тст (температуры стеклования) до Тт (температуры текучести). Если температурный интервал Тст–Тт достаточно широк и захватывает обычные температуры, то такие полимеры называют эластиками или эластомерами, или каучуками. Полимеры с узким интервалом температур Тст–Тт, смещенным в область повышенных температур, называют пластиками или пластомерами. При обычных температурах пластики находятся в стеклообразном состоянии. При температуре выше Тт (область III) полимер переходит в вязкотекучее состояние. Повышение температуры выше Тр ведет к деструкции, разрушению полимера. вещество в вязкотекучем состоянии под действием напряжений сдвига течёт как вязкая жидкость, причём деформация полимера является необратимой (пластической).
Свойства полимеров
Химические свойства полимеров зависят от состава, молекулярной массы и структуры. Полимерам свойственны реакции соединения макромолекул поперечными связями, взаимодействие функциональных групп друг с другом и низкомолекулярными веществами, деструкция. Наличие у макромолекул двойных связей и функциональных групп обуславливает повышение реакционной способности полимеров.
Вследствие наличия двойных связей и функциональных групп отдельные макромолекулы могут сшиваться поперечными связями, например, при вулканизации каучука происходит переход линейных макромолекул в сетчатую структуру резины (0,5-5% серы) или эбонита (более 20% серы).
К реакциям взаимодействия функциональных групп с низкомолекулярными веществами относятся галогенирование полиолефинов, гидролиз полиакрилатов и др. Полимеры могут подвергаться деструкции, то есть разрушению под действием кислорода, света, теплоты и радиации. В результате деструкции уменьшается молекулярная масса макромолекул, изменяются физические и химические свойства. Процесс ухудшения свойств полимеров во времени в результате деструкции называется старением и в конце концов полимеры становятся непригодными для дальнейшего применения. Для замедления деструкции вводят стабилизаторы, чаще всего антиоксиданты, то есть ингибиторы реакции окисления (фосфиты, фенолы, ароматические амины).
Механические свойства полимеров
Они определяются элементным составом, молекулярной массой, структурой и физическим состоянием макромолекулы.
Механическая прочность полимеров возрастает с увеличением их молекулярной массы, при переходе от линейных к разветвленным и далее к сетчатым структурам. Стереорегулярные структуры имеют более высокую прочность, чем полимеры с разупорядоченной структурой. Дальнейшее увеличение механической прочности полимеров наблюдается при их переходе в кристаллическое состояние. Механическая прочность может быть также повышена путём добавления наполнителей, например, сажи и мела, армированием волокнами (например, стекловолокном).
Электрические свойства полимеров
По этим свойствам все вещества подразделяются на диэлектрики, полупроводники и проводники.
Диэлектрики имеют очень низкую проводимость (σ<10-8 Ом-1см-1), которая увеличивается с повышением температуры. Большинство полимеров относится к диэлектрикам. Однако их диэлектрические свойства лежат в широких пределах и зависят от состава и структуры макромолекул, в значительной степени определяются наличием, характером и концентрацией полярных групп в макромолекулах. Наличие галогенных, гидроксильных карбоксидных и других полярных групп ухудшают диэлектрические свойства полимеров. Поэтому хорошими диэлектриками являются полимеры, не имеющие полярных групп, такие, как фторопласт, полиэтилен, полиизобутилен, полистирол. С увеличением молекулярной массы полимера улучшаются его диэлектрические свойства. При переходе к стеклообразному к высокоэластичному и вязкотекучему состояниям возрастает удельная электрическая проводимость.
Некоторые функциональные группы, например, гидроксильные, обуславливают гидрофильность полимеров. Такие полимеры поглощают воду, которая приводит к повышению электрической проводимости. Поэтому гидроксильные группы связывают между собой или с другими группами реакциями конденсации. Полимерные диэлектрики широко используют в электро- и радиотехнике как материалы различных электротехнических изделий, защитных покрытий кабелей, проводов, изоляционных эмалей и лаков.
К органическим полупроводникам относят вещества, электрическая проводимость которых лежит в пределах 10-10–10-3 Ом-1см-1. Электрическая проводимость полупроводников возрастает с увеличением температуры и при воздействии света. Некоторые полимеры с системой сопряженных двойных связей обладают полупроводниковыми свойствами, которые обусловлены наличием нелокализованных π-электронов сопряженных двойных связей. Примеры органических полупроводников: полиацетилен (-СН2=СН2-)n, поливинилены (-СН=СR-)n, полинитрилы (-N=CR-)n и другие. Электрическая проводимость может резко возрастать при химическом или электрохимическом окислении или восстановлении некоторых полимеров, например, полиацетилена, полианилина (-C6H3NH2-)n, полипиролла (-C4H3N-)n, других. При электрохимическом окислении в состав полимера внедряются анионы, например, ClO4–, при восстановлении – катионы (например, Li+). Такие органические полупроводники применяют в качестве электродных материалов аккумуляторов, пластин конденсаторов, материалов сенсоров, а в перспективе и для замены металлов (органические металлы).
Смесь некоторых полимеров, находящихся в аморфном состоянии, например, полиэтиленоксида (-СН2-СН2-О-)n с солями металлов, например,
LiClO4, обладает ионной проводимостью. Такие твёрдые электролиты могут получить применение в аккумуляторах.
Применение полимеров
На основе полимеров получают следующие материалы: волокна, плёнки, резины, лаки, клеи, пластмассы, композитные материалы (композиты).
Волокна получают путём продавливания растворов или расплавов полимеров через тонкие отверстия (фильеры) в пластине с последующим затвердеванием. К волокнообразующим полимерам относятся полиамиды, полиакрилонитрилы и другие.
Полимерные плёнки из расплавов полимеров используют в качестве электроизоляционного и упаковочного материала, основы магнитных лент и так далее.
Лаки – растворы плёнкообразующих веществ в органических растворителях. Кроме полимеров, лаки содержат вещества, повышающие пластичность (пластификаторы), растворимые красители, отвердители и др. Применяются для электроизоляционных покрытий, в качестве основы грунтовочного материала и лакокрасочных эмалей.
Клеи – композиции, способные соединять различные материалы за счёт образования прочных связей между поверхностями и клеевой прослойкой. Клеи подразделяют на термопластические, термореактивные и резиновые. В качестве полимерной основы термореактивных клеев служат фенол- и мочевиноформальдегидные и эпоксидные смолы, полиуретаны, полиэфиры и др., термопластичных – полиакрилы, полиамиды, поливинилацетали, поливинилхлорид и другие полимеры.
Пластмассы – это материалы, содержащие полимер, который при формировании изделия находится в вязкотекучем состоянии, а при его эксплуатации – в стеклообразном. Они подразделяются на термопласты и реактопласты. При формовании реактопластов происходит необратимая реакция отвердевания, к ним относятся материалы на основе фенолформальдегидных, мочевиноформальдегидных, эпоксидных и других смол. Термопласты способны многократно переходить в вязкотекучее состояние при нагревании, и стеклообразное – при охлаждении. К ним относятся материалы на основе полиэтилена, полипропилена, политетрафторэтилена, поливинилхлорида, полистирола и др. Кроме полимеров, в состав пластмасс входят пластификаторы (диоктилфталат, дибутилсебацинат, хлорированный парафин), снижающие температуру стеклования и повышающие текучесть полимеров; красители и наполнители (графит, сажа, мел, металл, бумага, ткань и др.), улучшающие физико-механические свойства полимеров.
Композиты – состоят из основы (органической, полимерной, углеродной, керамической, металлической), армированной наполнителем (высокопрочными волокнами или нитевидными кристаллами). Армирующие наполнители в значительной степени определяют механические, теплофизические и электрические свойства композитов. Композиты на основе полимеров используются как конструкционные, электро- и теплоизоляционные, коррозионностойкие, антифрикционные материалы в автомобильной, станкостроительной, электротехнической, авиационной, радиотехнической, горнорудной промышленности, космической технике, химическом машиностроении и строительстве.
В настоящее время широко применяется большое число различных полимеров. Физические и химические свойства некоторых термопластов приведены в таблице 14.2 и 14.3 [1].
ЛЕКЦИЯ ПО ТЕМЕ: «Методы химической идентификации веществ»
План
1. Единицы количества вещества: моль, молярная масса, молярный объём.
2. Способы выражения концентрации: массовая, молярная, мольная, нормальная.
3. Химическая идентификация веществ: качественный и количественный анализ, аналитический сигнал, инструментальные методы анализа; химический анализ
Единицы количества вещества
Количество вещества – это число структурных элементов (атомов, молекул, ионов) в системе. Единицей измерения вещества является моль, то есть количество вещества в системе, которое содержит столько определенных структурных элементов (молекул, атомов, ионов, электронов и др.), сколько атомов содержится в 0,012 кг углерода-12. Используя термин «моль», следует указывать частицы, к которым он относится: «моль молекул», «моль атомов», «моль электронов» и т.д. Масса одного моля вещества называется молярной массой М с единицей измерения кг/моль или г/моль. Моль любого газа при нормальных условиях (0оС и 1.033˙105 Па) занимает объём 22,416 л. Этот объём называется молярным.
Способы выражения концентрации приведены в главе «Растворы» [1].
Химическая идентификация веществ
Химическая идентификация (качественный анализ) и измерение (количественный анализ) являются предметом аналитической химии.
Химическая идентификация (обнаружение) – это установление вида и состояния фаз, молекул, атомов, ионов и других составных частей вещества на основе сопоставления экспериментальных и соответствующих справочных данных для известных веществ. Идентификация является целью качественного анализа. При идентификации обычно определяют комплекс свойств веществ: цвет, фазовое состояние, плотность, вязкость, температуры плавления и кипения, фазового перехода; растворимость, электродный потенциал, энергию ионизации и (или) другие. При анализе многокомпонентных систем используют универсальные приборы (спектрометры, спектрофотометры, хроматографы, полярографы и др.), снабженные компьютерами, в памяти которых имеется справочная химико-аналитическая информация. На базе этих универсальных установок создается автоматизированная система анализа и обработки информации.
В зависимости от вида определяемых частиц различают элементный, молекулярный, изотопный и фазовый анализы. Чаще используют первые два вида анализа.
В зависимости от массы сухого вещества или объёма раствора анализируемых веществ различают макрометод (0,5-10 г или 10-100 мл), полумикрометод (10-50 мг или 1-5 мл), микрометод (1-5 мг или 0,1-0,5 мл) и ультрамикрометод (ниже 1 мг или 0,1 мл) идентификации.
Качественный анализ характеризуют пределом обнаружения сухого вещества, т есть минимальным количеством надёжно идентифицируемого вещества, и предельной концентрацией раствора Сх, min. Эти две величины связаны:
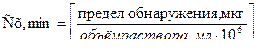
В качественном анализе используют только те реакции, пределы обнаружения которых меньше 50 мкг.
Иногда то или иное вещество можно обнаружить в присутствии других веществ с использованием специфических реакций. Чаще всего мешающие идентификации вещества переводят в осадок, слабодиссоциирующее или комплексное соединение, то есть используют маскирующие или отделяющие реакции.
Количественный анализ
Определение содержания (концентрации, массы и т.п.) компонентов в анализируемом веществе называется количественным анализом. С его помощью выявляют массовые соотношения компонентов в анализируемом образце, концентрацию вещества в растворе или газе. При количественном анализе измеряют те или иные химические, физико-химические и физические параметры анализируемого образца, которые зависят от его состава или содержания того или иного компонента. Результаты анализа обычно выражают в массовых долях, %.
Количественный анализ проводят в определенной последовательности, в которую входят отбор и подготовка проб, проведение анализа, обработка и расчёт результатов анализа. Как и в качественном анализе, различают макро-, полумикро-, микро- и ультрамикрометоды. Количественный анализ широко используют для изучения состава руд, металлов, неорганических и органических соединений. Особое внимание обращается на определение содержания токсичных веществ в воздухе, водоёмах, почвах, продуктах питания, различных товарах.
Аналитический сигнал
Практически все методы анализа основаны на зависимости каких-либо доступных измерению свойств вещества от их состава. Как правило, находят и используют уравнение связи между свойством и составом, разрабатывают способы регистрации количественных характеристик свойства, которые называют аналитическим сигналом. Величину аналитического сигнала переводят в единицы, характеризующие количество или концентрацию компонента. Измеряемыми величинами могут быть масса, объём, светопоглощение, электрический ток и т.д.
Ниже приведены названия методов и измеряемые величины.
| Измеряемая величина (свойство) | Название метода |
| Масса | Гравиметрический Масс-спектрометрический |
| Объём | Титриметрический Газоволюметрический |
| Плотность | Денсиметрический |
| Поглощение или испускание инфракрасных лучей | Инфракрасная спектроскопия |
| Колебания молекул | Комбинационное рассеяние |
| Поглощение или испускание видимых, ультрафиолетовых и рентгеновских лучей. Колебания атомов. Рассеяние света | Спектральный и рентгеноспектральный Фотометрический (колориметрия, спектрофотометрия идр.) Атомно-адсорбционная спектроскопия Люминесцентный анализ |
| Диффузионный ток на электроде | Полярография и вольтамперометрия |
| Электродный потенциал | Потенциометрический |
| Количество электричества | Кулонометрический |
| Электрическая проводимость | Кондуктометрический |
| Радиоактивность | Радиоактивных индикаторов |
| Скорость реакции | Кинетический Каталитический |
| Тепловой эффект реакции | Термометрия и калориметрия |
| Вязкость | Вискозиметрия |
| Поверхностное натяжение | Тензометрия |
| Понижение температуры замерзания | Криоскопия |
| Повышение температуры кипения | Эбуллиоскопия |
| Различная сорбция-десорбция | Хроматографический |
Химический анализ
Все методы количественного анализа делят на химические, физико-химические и физические. Это деление условно. К химическому анализу относятся гравиметрический, титриметрический, комплексонометрическое и окислительно-восстановительное титрование.
Гравиметрический метод. Сущность метода заключается в получении труднорастворимого соединения, в которое входит определяемый компонент. Затем, после отфильтровывания осадка, его высушивают, прокаливают и взвешивают. По массе вещества определяют массу нужного компонента, и проводят расчёт его массовой доли в анализируемой навеске. Имеются разновидности гравиметрического метода. Например, анализируемый компонент выделяют в виде газа, который взаимодействует с реактивом. По изменению массы реактива судят о содержании определяемого компонента в навеске. Например, СО32- + 2Н+ = Н2СО3 = Н2О + СО2, количество выделившегося углекислого газа можно определить по изменению массы вещества, например, СаО, с которым реагирует СО2. Гравиметрический метод трудоёмок и длителен.
Титриметрический анализ. Метод заключается в измерении объёма раствора, израсходованного на реакцию с анализируемым компонентом. Для этих целей используют титрованные растворы, то есть растворы с известной концентрацией – титром раствора. Определение проводят способом титрования, то есть постепенного приливания титрованного раствора к раствору анализируемого вещества, объём которого точно измерен. Титруют до достижения точки эквивалентности. Существует несколько разновидностей анализа: кислотно-основное, осадительное, комплексонометрическое, окислительно-восстановительное титрование.
Инструментальные методы анализа
Инструментальные методы анализа имеют массу достоинств: быстроту, высокую чувствительность, возможность одновременного определения нескольких компонентов, сочетание нескольких методов, автоматизация и использование компьютеров для обработки результатов анализа. Как правило, в инструментальных методах применяют сенсоры (датчики). Инструментальных методов очень много, мы рассмотрим только некоторые из них.
Электрохимические методы: потенциометрический, полярографический, кондуктометрический и др.
Потенциометрический метод основан на измерении электродных потенциалов, которые зависят от активности ионов, а в разбавленных растворах – от их концентрации. В основе лежит уравнение Нернста: 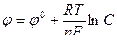 . Измерительная ячейка состоит из измерительного (индикаторного) электрода и электрода сравнения, который не чувствителен к определяемому веществу. Наиболее часто используют ионоселективные электроды, на границах раздела фаз которых протекают ионообменные реакции. Например, для измерения рН ионоселективным является стеклянный электрод. Существуют также ионоселективные электроды для определения концентрации ионов натрия, калия, аммония, хлора, кальция, магния, нитрат-анионов и т.д.
. Измерительная ячейка состоит из измерительного (индикаторного) электрода и электрода сравнения, который не чувствителен к определяемому веществу. Наиболее часто используют ионоселективные электроды, на границах раздела фаз которых протекают ионообменные реакции. Например, для измерения рН ионоселективным является стеклянный электрод. Существуют также ионоселективные электроды для определения концентрации ионов натрия, калия, аммония, хлора, кальция, магния, нитрат-анионов и т.д.
Кондуктометрический метод основан на измерении электрической проводимости разбавленных растворов, которая пропорциональна концентрации электролитов. Этим методам, например, определяют общее содержание примесей в воде высокой чистоты.
Хроматографический анализ позволяет разделять двух- и многокомпонентные смеси газов, жидкостей и растворенных веществ методом сорбции в динамических условиях. Основной прибор – хроматограф. Разработано несколько методов, которые классифицируют по механизму процесса и природе частиц – молекулярная, ионообменная, осадительная, распределительная хроматография; и по формам применения – колоночная, капиллярная, тонкослойная и бумажная. Молекулярная хроматография основана на различной адсорбируемости молекул на сорбентах (адсорбентах), ионообменная – на различной способности к обмену ионов раствора. В осадительной хроматографии используют различную растворимость осадков, а в распределительной – различное распределение веществ между двумя несмешивающимися жидкостями. К достоинствам метода следует отнести быстроту и надёжность, возможность определения нескольких компонентов смеси из раствора.
Оптические методы анализа основаны на измерении оптических свойств веществ и излучений, взаимодействии электромагнитного излучения с атомами или молекулами анализируемого вещества, вызывающего излучение, поглощение или отражение лучей. Они включают в себя эмиссионные, люминесцентные и абсорбционные спектральные методы.
Методы, основанные на изучении спектров излучения, называют эмиссионными спектральными методами. В эмиссионной спектроскопии проба вещества нагревается до очень высоких температур (2000-15000 оС). Вещество, испаряясь, диссоциирует на атомы или ионы, которые дают излучение. В спектрографе излучение разлагается на спектр цветных линий. Сравнение этого спектра со справочными данными позволяет определить вид элемента, а интенсивность спектральных линий – его количество. Преимущества этого метода: быстрота выполнения анализа, возможность определения нескольких компонентов из одной навески.
Разновидность эмиссионного анализа – эмиссионная пламенная фотометрия, в которой исследуемый раствор вводят в бесцветное пламя горелки. По изменению цвета пламени определяют вид вещества, а по интенсивности окраски – о его концентрации. Метод выполняют с помощью пламенного фотометра и используют для анализа щелочных, щелочноземельных металлов и магния.
Методы, основанные на свечении анализируемого вещества ультрафиолетовых (фотолюминесценция), рентгеновских (рентгенолюминесценция) и радиоактивных (радиолюминесценция) лучей называют люминесцентными. Люминесцентные методы обладают очень высокой чувствительностью (до 10-10-10-13 г люминесцирующих примесей).
Методы, основанные на изучении спектров поглощения лучей анализируемыми веществами, называют абсорбционно-спектральными. При прохождении света через раствор свет или его компоненты поглощаются или отражаются. По величине поглощения или отражения лучей судят о природе и концентрации вещества. Определение ведут с помощью спектрофотометров или фотоколориметров. Если измеряют поглощение лучей атомами определяемого компонента, которые получают распылением раствора анализируемого вещества в пламени горелки, то метод называют атомно-абсорбционным. Оптический метод, основанный на отражении света твёрдыми частицами, взвешенными в растворе, называется нефелометрией, а прибор, используемый в нём – нефелометром.
Таким образом, использование законов электрохимии, сорбции, эмиссии, поглощения или отражения излучения и взаимодействия частиц с магнитными полями позволило создать большое число инструментальных методов анализа, характеризуемых высокой точностью, чувствительностью, быстротой и надёжностью определения, возможностью анализа многокомпонентных систем.
Заключение
Вопросы, рассмотренные в курсе лекций, позволяют получить современное научное представление о материи, формах ее движения, веществе как одном из видов материи, механизме превращения химических соединений, свойствах технических материалов и применении химических процессов в современной технике. Понимание химических законов помогает инженеру в решении технических проблем. Знание химии необходимо для последующего успешного изучения общенаучных и специальных дисциплин, таких, как сопротивление материалов, материаловедение, основы теплопередачи, электротехника, электроника, энергетика, а также ряда дисциплин, связанных с железнодорожным транспортом.
Знание химии полезно будущему инженеру, так как он будет постоянно сталкиваться с различными веществами и процессами. Научно-технический прогресс вызывает к жизни все новые материалы, новые машины, аппараты и приборы, в которых широко используются достижения химии.
Конспект курса лекций будет полезен при изучении, а также повторении материала в период подготовки к экзамену.
Литература
1. Коровин Н.В. Общая химия. – М.: Высшая шк., 2000.
2. Зубрев Н.И. Инженерная химия на ж/д транспорте. – М.: 1999
3. Жук Н.П. Курс теории коррозии и защиты металлов. – М.:2002
4. Глинка Н.Л. Задачи и упражнения по общей химии. – М.:1998
5. Глинка Н.Л. Общая химия. – М.: Интеграл-пресс, 2003.
Дата: 2018-12-21, просмотров: 728.