Бактериальная клетка состоит из клеточной стенки, цитоплазматической мембраны, цитоплазмы с включениями и нуклеотида. Дополнительные структуры: капсула, микрокапсула, жгутики, реснички, плазмиды, споры и т.д.
Клеточная стенка – прочная, упругая, придаёт бактериям определённую форму, сдерживает высокое осмотическое давление в бактериальной клетке. Толщина клеточной стенки у грациликут 15-20 нм, у фирмикут – 50 нм (т.е. у фирмикут клеточная стенка в 2-4 раза толще).
В клеточной стенке фирмикут содержится небольшое количество полисахаридов, липидов, белков. Большая часть массы (40-90%) клеточной стенки этих микроорганизмов составляет пептидогликан (муреин, мукопептид), ковалентно связанный с тейхоевыми кислотами (от греч. teichos – стенка), молекулы которых представляют собой цепи из 8-50 остатков глицерола и рибитола, соединенных фосфатными мостиками.
Пептидогликан представлен параллельно распложенными молекулами гликана, состоящего из остатков N-ацетилглюкозамина и N-ацетилмурамовой кислоты, соединенных гликозидной связью, типа b (1à4). Лизоцим, являясь ацетилмурамидазой, разрывает эти связи.
Гликановые молекулы соединены пептидной связью, поэтому этот полимер назван пептидогликан.
Элементы пептидогликана являются отличительными особенностями бактерий и отсутствуют у человека.
Способность фирмикут при окраске по Грамму удерживать генциановый виолетовый в комплексе с йодом (сине-фиолетовая окраска бактерий) связана со свойством многослойного пептидогликана взаимодействовать с красителем. Кроме того, последующая обработка спиртом вызывает сжимание пор в пептидогликане и задержку красителя в клеточной стенке.
В клеточной стенке грациликут пептидогликана содержится меньше (5-10%). В состав их клеточной стенки входит наружная мембрана, связывающаяся посредством липопротеина с подлежащим слоем пептидогликана.
Наружная мембрана представляет собой волнообразную трёхслойную структуру, сходную с внутренней мембраной, которую называют цитоплазматической. Основным компонентом этих мембран служит билипидный слой.
Наружная мембрана является ассиметричной мозаичной структурой, представленной липополисахаридами, фосфолипидами и белками.
С её внешней стороны расположен липополисахарид, состоящий из 3-х компонентов:
1. Липид А;
2. Стержневая часть или ядро;
3. О-специфическая часть (образована повторяющимися олигосахаридными последовательностями).
 ЛПС «заякорён» в наружной мембране липоидом А, обуславливающем токсичность ЛПС и отождествляемому с эндотоксином. О-антиген определяет серогруппу, серовар бактерии.
ЛПС «заякорён» в наружной мембране липоидом А, обуславливающем токсичность ЛПС и отождествляемому с эндотоксином. О-антиген определяет серогруппу, серовар бактерии.
Экзотоксины
Экзотоксины – белки, представляющие биофункциональную структуру, т.к. они имеют транспортную группу, которая взаимодействует со специфическим рецептором клетки и токсическую группу (активатор), которая проникает внутрь клетки и блокирует жизненно важные процессы.
Экзотоксины делят на 3 класса:
Ø Класс А. Токсины, секретирующиеся во внешнюю среду (гистотоксин Corynebacterium diphteriae; отёчный, летальный токсин Bacillus anthracis);
Ø Класс В. Токсины, частично секретируемые и частично связанные с микробной клеткой (тетаноспазмин Clostridium tetani, нейротоксин Clostridium botulinum);
Ø Класс С. Токсины, связанные с микробной клеткой и попадающие в окружающую среду при аутолизе клетки (цито-, энтеро-, нейротоксин Shygella dysenteriae, «мышиный» токсин Yersinia pestis).
Механизм действия белковых токсинов сводится к повышению проницаемости мембран эритроцитов, лейкоцитов и других клеток (мембранотоксины) или к блокаде синтеза белка и других биохимических процессов в клетках (цито-, энтеро- и нейротоксины), либо нарушению взаимосвязи и взаимодействия клеток.
Эндотоксины
Эндотоксины – белково-липополисахаридные комплексы клеточной стенки грациликут, которые выделяются в окружающую среду при лизисе бактерий.
Эндотоксины термостабильны, менее ядовиты, чем экзотоксины, действуют быстро, но не обладают специфичностью действия, малочувствительны к химическим веществам, не переходят в анатоксин.
Антитела, образующиеся к О-специфическим цепям ЛПС, не нейтрализуют их токсическое действие.
Основной точной приложения действия эндотоксинов являются макрофаги, которые в ответ на их действие выделяют эндогенные пирогенны. Кроме того, эндотоксины способны активировать комплемент по альтернативному пути.
Эндотоксиновый шок наиболее демонстративен при менингококковой инфекции. Характерным является появление симптомов шока или усиление их после применения бактерицидных антибиотиков, что связано с интенсивным бактериолизом и выбросом эндотоксинов. Данная реакция встречается и при другой этиологии ИТШ и также при инфекциях, протекающих без явлений шока. Например, с этой реакцией сталкиваются венерологи при лечении вторичного свежего сифилиса. После первых инъекций пенициллина у больных отмечается повышение температуры тела и усиление воспаления в области сифилид – розеолы приобретают более насыщенный розово-красный цвет, становятся хорошо видны, как бы «подкрашиваются». Это связано с интенсивным лизисом бледной спирохеты и усилением иммунных реакций на продукты распада.
Данный тип реакций называется реакцией обострения Герксгеймера-Яриша-Лукашевича или реакцией бактериолиза. Он подтверждает участие в патогенезе шока продуктов распада бактериальных клеток (в случае менингококковой инфекции – менингококкового эндотоксина).
Таким образом, в некоторых случаях при развитии ИТШ и высоком риске участия грациликут в качестве этиологического фактора, предпочтение может быть отдано не бактерицидным, а бактериостатическим антибиотикам.
Патогенез
Инфекционно-токсический шок (син. циркуляторный, септический, грампозитивный, грамнегативный, эндотоксиновый, экзотоксиновый) - развивается в результате воздействия эндотоксинов и бактериальных продуктов на клеточные мембраны, компоненты свертывания крови и комплемент, что приводит к повышению свертываемости, повреждению клеток и нарушению кровотока, особенно микроциркуляции.
Система комплемента состоит не менее чем из 20 различных самоустанавливающихся протеинов и может быть активирована каким-либо одним из по меньшей мере двух пусковых факторов. Во время активации ранее синтезированные биологически активные протеины превращаются в гуморальные медиаторы воспаления и альтерации тканей. Активация комплемента происходит ступенчато, наподобие каскада свертывания крови. Образование полного комплемента приводит к лизису мембраны клеток бактерий, эритроцитов и других тканей. Высвободившиеся во время активации комплемента фрагменты пептидов активируют другие клеточные и гуморальные эффекторные системы. Известны два пути активации системы комплемента: классический и альтернативный. Альтернативная активация (называемая также пропердиновой) может произойти под влиянием неиммунологических факторов, независимо от антител. В литературе имеются данные об активации комплемента липополисахаридами (эндотоксины). Активация комплемента приводит к образованию низкомолекулярных пептидов - факторов комплемента С3,С4 и С5, опосредующих клеточные и гуморальные реакции. Фрагменты С3а, С4а и С5а называют анафилотоксинами. Они стимулируют высвобождение гистамина из тучных клеток и базофилов, вызывают сокращение гладких мышц и увеличивают проницаемость сосудов. Считается, что фрагмент С2 обладает кининовой активностью, вызывая увеличение проницаемости сосудов. Фрагмент С5а, взаимодействуя со специфическими высокоаффинными рецепторами гранулоцитов и тромбоцитов, вызывает агрегацию клеток, усиление прилипания, хемотаксис и активацию клеток. Активированные таким образом нейтрофилы высвобождают метаболиты арахидоновой кислоты, бескислородные радикалы и лизосомальные ферменты, вызывающие воспалительные изменения в тканях и увеличивающие проницаемость капилляров. Данный механизм может иметь определенное значение в возникновении дыхательной недостаточности и вазодилатации при септическом состоянии, вызванном грамотрицательными микроорганизмами.
Литературные данные, полученные в последнее время свидетельствуют также о том, что под влиянием эндотоксинов и других бактериальных продуктов выделяются эндогенные цитокины, основными мишенями для которых являются лейкоциты, эндотелий и сердце. Появляющиеся медиаторы воспаления и сами эндогенные цитокины оказывают большое воздействие на вазомоторный тонус, проницаемость мелких сосудов и агрегацию лейкоцитов и тромбоцитов. Происходит перестройка в терминальном отделе системы кровообращения. В результате этого возникает утрата тонуса как сосудов сопротивления (артериальных), так и объемных (венозных). Кровь может скапливаться в капиллярном русле, а белки плазмы пропотевают в интерстициальную жидкость. В венозной системе также отмечается депонирование крови. В результате стимуляции b-рецепторов открываются артериовенозные шунты конечной части кровотока.
Достаточно важное значение в патогенезе инфекционно-токсического шока в настоящее время придается также образованию в организме нитратов. При воспалительной реакции в организме ключевую роль в образовании нитратов играют макрофаги. Специфический фермент макрофагов - NO-синтаза (макрофагальная, которая локализуется в макрофагах, миокарде и гладкой мускулатуре) превращает аргинин в NO, из которого затем могут образовываться нитриты и нитраты. Главная функция NO, который синтезируется макрофагами, состоит в обеспечении их цитотоксического действия. При активации бактериальными эндотоксинами или Т-лимфоцитами макрофаги усиливают синтез NО-синтазы, которая превращает аргинин в NO. Выделяясь из макрофагов, NO быстро проникает в бактерии и клетка погибает. Таким образом, NO играет важную роль в иммунной защите организма. Кроме того NO способствует снижению активности пограничных воспалительных клеток, тормозит агрегацию тромбоцитов и улучшает местное кровообращение. Патогенное же влияние образования NO в организме при воспалении может заключаться в следующем. При воспалительных процессах в организме могут образовываться активные формы кислорода, которые являются одной из важных молекулярных мишеней для NO. NO связывается с кислородом, образуя пироксинитриты, по токсичности во много раз превосходящие NO. Они то и играют важную роль во многих патофизиологических процессах, включая септический шок, а также ишемические и язвенные поражения органов. Пироксинитрит вызывает повреждение белков и липидов клеточных мембран, повреждает сосудистый эндотелий, увеличивает агрегацию тромбоцитов, участвует в процессах эндотоксемии. Сама NO, избыточно накапливаясь в клетке, может вызывать повреждение ДНК и обладать провоспалительным действием при септическом шоке.
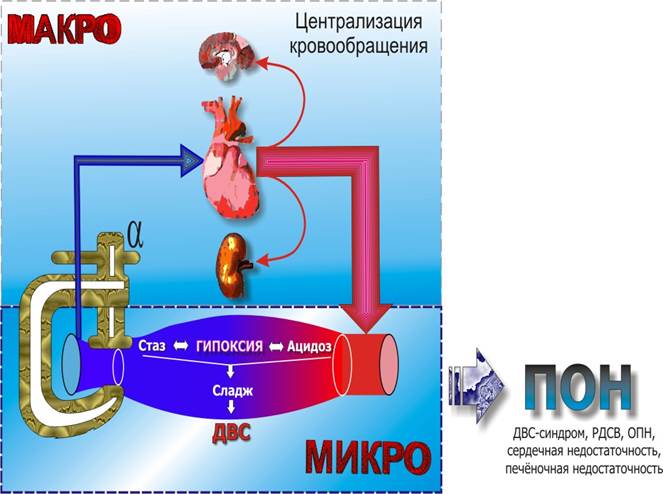
Таким образом, на начальном этапе развития заболевания под воздействием эндотоксинов происходит в первую очередь расширение стенок мелких сосудов (в основном венул), а также значительно повышается проницаемость сосудистой стенки. В результате всего вышеизложенного, несмотря на отсутствие абсолютного дефицита объема, венозный возврат к сердцу уменьшается (относительная гиповолемия). В ответ на это происходит рефлекторное симпатическое сужение вен. Но активное сужение вен эффективно уменьшает венозное кровенаполнение только в том случае, если вены хорошо наполнены и растянуты. Если же трансмуральное давление достаточно низко, чтобы привести вены в полуспавшееся состояние, даже сильные сокращения гладкой мышцы вен оказывают только незначительное влияние на количество крови в них. При такой ситуации сужение вен может даже несколько увеличить местную емкость вен, так как оно делает стенку более жесткой, в результате чего просвет становится большим и приобретает более круглую форму, несмотря на то, что просвет окружности уменьшается. В результате снижения венозного возврата повышается активность симпатической нервной системы, что наряду с непосредственным влиянием эндотоксинов ведет к сокращению пре- и посткапиллярных сфинктеров (стимуляция a-рецепторов). В результате кровоснабжение тканей становится недостаточным, минутный объем сердца в эту фазу большей частью нормальный или даже повышенный (т. е. МОС нормальный или увеличенный, ЧСС увеличивается, общее периферическое сопротивление уменьшается и снижается АД). Артерио-венозная разница по кислороду и обеспечение периферии кислородом понижены.
В дальнейшем по мере дальнейшего развития шока происходит формирование рокового порочного круга. Прекапиллярные артериальные сфинктеры более чувствительны к токсическим влияниям (в том числе ацидозу), поэтому их спазм быстро сменяется парезом. Посткапиллярные (венулярные) сфинктеры более устойчивы к метаболическим нарушениям и длительное время остаются в состоянии тонического напряжения. Таким образом, кровь, притекающая в капиллярное русло депонируется, в связи с чем нарастает тканевая гипоксия, усугубляется метаболический ацидоз, пропотевает плазма с нарастающей компрессией капилляров, что наряду с застоем крови в венозном русле, способствует дальнейшему уменьшению венозного возврата и увеличению относительной гиповолемии.
В результате возникает следующая причинно-следственная связь: стаз в капиллярах - висцеральный застой - уход воды - повышение вязкости крови - агрегация красных и белых кровяных телец, образование красного и белого тромба - истощение факторов свертывания и тромбоцитов вследствие диссеминированного внутрисосудистого свертывания - возникновение изнуряющей коагулопатии с повышенной предрасположенностью к кровотечению.
В пораженных областях аэробные энергетические пути переключаются на анаэробный гликолиз. Переключение окислительного обмена на гликолитический путь значительно увеличивает потребление глюкозы при одновременном уменьшении выхода АТФ. Это снова ведет к снижению уровня глюкозы. Биосинтез белка при шоке ограничен. Это особенно быстро влияет на синтез тех белков, которые имеют короткое время полужизни, например, факторы свертывания. Таким образом, нарушение свертывания крови еще более усиливается. При шоке начинается выход калия из клеток. Метаболический ацидоз возникает в результате увеличенной продукции лактата, а также пирувата, a-кетоглутарата и кетоновых тел. Ацидоз частично компенсируется усиленным дыханием. В результате повышения концентрации Н+ в плазме наблюдается следующее: отрицательное инотропное действие на сердце; снижение чувствительности прекапиллярных сфинктеров в смысле вазомоторных реакций с образованием отеков; повышение выброса катехоламинов; активирование свертывающей системы в качестве одной из причин диссеминированного внутрисосудистого свертывания. Некоторые вещества, образующиеся при шоке (в частности и сами цитокины), обладают отрицательным инотропным действием. Длительное действие этих факторов ведет к расширению сердца и сердечной недостаточности, тем самым, к уменьшению МОК.
Почки. По причине эфферентного сокращения сосудов при шоке уменьшается давление гломерулярной фильтрации, в результате чего развивается олигурия (4-20 мл/ч) или анурия (4 мл/ч). Сужение почечных сосудов сохраняется еще долгое время после нормализации давления крови. Ишемия вызывает прогрессирующий некроз канальцев вследствие гломерулярной, а затем тубулярной недостаточности с образованием цилиндров в дистальных канальцах. Признаком почечной недостаточности является увеличение содержания в крови таких, обычно выделяющихся с мочой соединений, как мочевина и креатинин.
Таким образом, одним из основных патофизиологических механизмов в развитии инфекционно-токсического шока при пневмониях следует считать развитие гиповолемии вследствие секвестрации крови в микроциркуляторном русле и выхода ее в ткани из-за повышения проницаемости капилляров. По образному выражению И. Теодореску-Ексари при ИТШ происходит «кровотечение в собственные сосуды».
Патогенетические стадии
Стадия компенсации
Компенсаторные прессорные реакции, направленные на поддержание АД и восполнение ОЦК. Происходит выброс катехоламинов, вызывающих спазм прекапиллярных сфинктеров. Открываются прямые артериовенозные шунты.
Стадия субкомпенсации
Инициальный спазм прекапилляров сменяется парезом и застоем крови в вено-венулярном отделе микроциркуляторного русла. В эту стадию развивается циркуляторная недостаточность (шок). Снижается венозный возврат к сердцу à снижается сердечный выброс. Включаются компенсаторные реакции – тахикардия, резорбция провизорного фильтрата в почечных канальцах. При уменьшении венозного возврата на 25-30% начинается декомпенсация со снижением АД и нарушением перфузии жизненно важных органов. В этих условиях реализуются механизмы централизации кровообращения.
Нарастает метаболический ацидоз à тахипное. возникают биоплярные изменения КОС – респираторный алкалоз в малом круге и метаболический ацидоз в большом.
Стадия декомпенсации
Характеризуется присоединением ДВС-синдрома à тканевая гипоксия à изменения в шоковых органах.
«Предтечей» ДВС синдрома является изменение реологии крови – сладж феномен.
В нормальных условиях кровь имеет характер стабильной суспензии.
Сохранность суспензионной стабильности крови обеспечивается величиной отрицательного заряда эритроцитов и тромбоцитов (дзета-потенциал – потенциал поверхности скольжения частицы в коллоидном растворе), определяемом соотношением белковых фракций (альбумины/глобулины) и достаточностью кровотока.
Уменьшение отрицательного заряда эритроцитов (увеличение глобулинов или фибриногена, их абсорбция на поверхности эритроцитов) приводит к развитию сладжирования крови.
Сладж (от англ. sludge – густая грязь, тина) – изменение реологии крови, характеризующееся прилипанием друг к другу эритроцитов, лейкоцитов и тромбоцитов и повышением вязкости крови, что затрудняет её перфузию через микрососуды.
Сладж может быть обратимым (только при агрегации эритроцитов) и необратимым (агглютинация эритроцитов).
Типы сладжа:
1. Классический – крупные размеры агрегатов, неровные очертания контуров и плотная упаковка эритроцитов;
2. Декстрановый – различная форма и величина, округлые очертания, плотная упаковка;
3. Аморфный грануловидный – мелкие агрегаты в виде гранул, состоящих из нескольких эритроцитов.
Сладж à резкое замедление кровотока, сепарация плазмы от эритроцитов, маятникообразные движения плазмы à стаз.
В связи с закупоркой терминальных артериол большим количеством агрегатов, капиллярные сосуды пропускают только плазму. При этом повреждается стенка сосудов (набухание, десквамация эндотелия).
Параллельно с процессами сладжирования и предшествуя им происходит нарушение проницаемости сосудов обмена (капилляры, венулы) в сторону её повышения. Это приводит к выходу жидкой части крови в интерстиций и сгущению крови, что усиливает процессы агрегации эритроцитов.
Морфологической основой проницаемости сосудов является эндотелий и базальная мембрана + периваскулярная ткань = неспецифический гистогематический барьер.
В морфологическом отношении повышение сосудистой проницаемости характеризуется увеличением промежутков между эндотелиоцитами вследствие сокращения и усиления везикулярного транспорта.
Раннее повышение проницаемости связано с воздействием биологически активных аминов и брадикинина, позднее (более 60 минут) – протеаз, каллидина, глобулярных веществ нейтрофилов. Действие этих веществ направлено на стенку капиллярных сосудов – межклеточный цемент эндотелия и базальную мембрану и заключается в физико-химических изменениях (деполимеризация сложных белково-полисахаридных комплексов).
Схема патогенеза шока
Клиническая картина
Шок развивается на фоне нарастания интоксикации: у больного появляется озноб, за которым следует резкий подъем температуры, часто наблюдается тошнота, рвота, диарея, состояние прострации.
В клиническом плане выделяют следующие группы симптомов, определяющих степень тяжести и прогноз.
| № | Критерии септического синдрома |
| 1. | Клинические признаки инфекции |
| 2. | Температура тела выше 38о С или ниже 36о С |
| 3. | Частота дыхания > 20 в минуту |
| 4. | Число сердечных сокращений > 90 в минуту |
| 5. | Количество лейкоцитов больше 12,0х109/л или меньше 4,0х109 /л или при 10% незрелых форм |
| 6. | 1 или более изменений со стороны следующих органов: - нарушение сознания; - Ро2 < 70 мм рт. ст.; - олигоурия < 30 мл/час; - увеличение уровня лактата. |
К числу ранних признаков начинающегося ИТШ относятся гипервентиляция, вызывающая респираторный алкалоз, и церебральные нарушения в виде беспокойства или заторможенности. Эти первые симптомы шока часто не привлекают внимание, что приводит к запоздалой диагностике и ухудшает прогноз. По мере развития болезни усиливается тахикардия, одышка, артериальная гипотензия, а иногда тенденция к гипертензии, наблюдается бледность конечностей с акроцианозом. Кожные покровы теплые и сухие («теплый шок»).
При исследовании в этот период определяется: снижение ОПСС и могут появляться ранние признаки снижения фракции выброса (нормализуется на 7-10 сутки), увеличение сердечного выброса, ЧСС, частоты дыханий и Ро2 смешанной венозной крови (из-за артериовенозного сброса)
При прогрессировании шока развивается артериальная гипотензия, нарастает олигурия. При исследовании у этих больных отмечаются низкие значения центрального венозного давления (ЦВД), низкий объем циркулирующей крови (ОЦК) и сердечный выброс, отмечается повышение ОПСС, ЛСС, альвеолярно-артериального градиента Ро2, уменьшение рН и Ро2 в артериальной крови т. е. гиподинамическая реакция системного кровообращения, нарастает олигурия и молочнокислая ацидемия. Возникает полиорганная недостаточность (СН, острая почечная недостаточность, РДСВ, печеночная недостаточность, ДВС-синдром).
Дата: 2019-07-30, просмотров: 466.