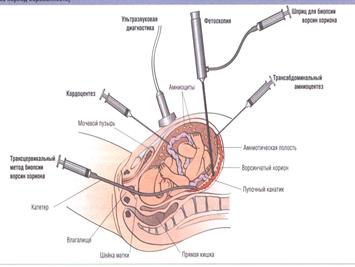
Это неинвазивный метод дородовой диагностики некоторых тяжелых заболеваний у плода. За рубежом его часто называют "тройным" тестом, поскольку при его проведении исследуется содержание в крови беременной женщины трех веществ: альфа-фетопротеина (АФП), хорионического гонадотропина (ХГ) и неконъюгированного эстриола (НЭ).
Кровь для исследования чаще всего берется из локтевой вены будущей мамы дважды: на сроке 15 недель и через 1-3 недели с таким расчетом, чтобы второй забор крови был не позже 20 недели беременности.
Полученный материал исследуют цитогенетически, биохимически, методами ДНК-диагностики. Врач сообщает семье результаты.
По результатам (семья) сама беременная женщина принимает решение о продолжении или прерывании беременности.
Для подавляющего большинства наследственных болезней эффективных способов лечения не существует. Из этого следует, что в борьбе с наследственной патологией основная роль отводится профилактике рождения аномального потомства. Общий профилактический характер носят мероприятия, направленные на оздоровление окружающей среды, способствующие снижению ее мутагенного воздействия на наследственный материал человеческого организма. В последние десятилетия распространенным и эффективным способом профилактики наследственных болезней является медико-генетическое консультирование.
Медико-генетическое консультирование — это один из видов специализированной помощи населению, направленной в первую очередь на предупреждение появления в семье детей с наследственной патологией. С этой целью составляют прогноз рождения в данной семье ребенка с наследственной болезнью, родителям объясняют вероятность этого события и оказывают помощь в принятии решения. В случае большой вероятности рождения больного ребенка родителям рекомендуют либо воздержаться от деторождения, либо провести пренатальную диагностику, если она возможна при данном виде патологии.
Консультирование семей, обращающихся к врачу-генетику, включает три основных этапа. Как правило, за консультацией обращаются семьи, где уже имеется ребенок с наследственной патологией, или семьи, в которых имеются больные родственники. На первом этапе консультирования производится уточнение диагноза, что является необходимой предпосылкой любого консультирования. Уточнение диагноза в медико-генетической консультации проводят с помощью генетического анализа. Для этой цели используют генеалогический, цитогенетический, биохимический и другие требуемые методы исследований, которым подвергаются пробанд и его родственники. Точный клинический и генетический диагноз заболевания позволяет установить степень генетического риска и выбор эффективных методов пренатальной диагностики и профилактического лечения.
На втором этапе консультирования делают прогноз потомства. Генетический риск может быть определен либо путем теоретических расчетов, основанных на генетических закономерностях, либо с помощью эмпирических данных. Сущность генетического прогноза заключается в определении вероятности появления наследственной патологии в семье. Наиболее эффективным является проспективное консультирование, когда риск рождения больного ребенка определяют до наступления беременности или в ранние ее сроки. Такие консультации чаще проводят в случае кровного родства супругов, при отягощенной наследственности по линии мужа или жены, при воздействии вредных средовых факторов на супругов незадолго до наступления беременности. Ретроспективное консультирование проводят после рождения больного ребенка относительно здоровья будущих детей.
Определение прогноза потомства при разных формах наследственной патологии различно. При моногенных, менделирующих болезнях прогноз основывается на расчете вероятности появления потомства в соответствии с генетическими закономерностями. При этом, если известен тип наследования данного заболевания и по родословной удается установить генотип родителей, оценка риска сводится к анализу мевделевского расщепления. Если у пробанда установлена вновь возникшая мутация, то повторный риск рождения ребенка с такой же патологией незначителен.
Расчет риска при моногенном заболевании может осложниться при пониженной экспрессивности или неполной пенетрантности гена, позднем проявлении генетической аномалии, генетической гетерогенности заболевания и вообще в случае неточного диагноза.
При хромосомных болезнях определение риска повторного рождения потомства с хромосомными аномалиями зависит от того, нормальны ли кариотипы родителей, не обнаружено ли у них мозаицизма, не наблюдается ли семейной формы структурных аномалий хромосом. В случае отсутствия нарушений в кариотипе родителей вероятность повторного рождения второго ребенка с хромосомной аномалией оценивается по эмпирическим данным для каждого вида аномалии с учетом возраста родителей.
При мультифакториальных заболеваниях, т.е. заболеваниях с наследственным предрасположением, основой оценки риска являются эмпирические данные о популяционной и семейной частоте каждого из них.
Специфический генетический риск до 5% принято считать низким, до 10% — повышенным в легкой степени, до 20% —средним, выше 20% — высоким. Генетический риск средней степени расценивают как противопоказание к зачатию или показание к прерыванию уже имеющейся беременности. Возможность проведения пренатальной диагностики является определяющей для принятия положительного решения в отношении завершения беременности.
На третьем этапе консультирования врач-генетик в доступной форме объясняет семье степень генетического риска рождения наследственно аномального потомства, сущность пренатальной диагностики и помогает принять правильное решение в отношении деторождения. Однако окончательное решение этого вопроса остается за родителями.
Широкое использование медико-генетического консультирования, разработка способов пренатальной диагностики наследственных заболеваний позволяют существенно уменьшить вероятность появления потомства с наследственной патологией в отдельных семьях.
Жизнь представлена отдельными видами, являющимися совокупностями организмов, которые обладают свойствами наследственности и изменчивости. Эти свойства становятся основой эволюционного процесса.
Изменчивость — это способность дочерних организмов отличаться от родительских форм. Различают две основные формы изменчивости: фенотипическую (ненаследственную) и генотипическую (наследственную). Фенотипической или модификационной изменчивостью называют изменения фенотипа под действием факторов внешней среды без изменения генотипа. Так как при этом генотип не затрагивается, то эти изменения не наследуются.
В генетической информации организма заложена способность развития определенных свойств и признаков. Эта способность реализуется лишь в конкретных условиях среды. Генотипическая изменчивость — это изменчивость, связанная с изменением генотипа. Она подразделяется на комбинативную и мутационную. Комбинативная изменчивость связана с получением новых комбинаций имеющихся в генотипе генов. Мутационная изменчивость — это скачкообразное и устойчивое изменение генетического материала, передающееся по наследству.
97. Изменчивость как свойство, обеспечивающее возможность существования живых систем в различных состояниях.
Продолжительное существование живой природы во времени на фоне меняющихся условий было бы невозможным, если бы живые системы не обладали способностью к приобретению и сохранению некоторых изменений, полезных в новых условиях среды. Свойство живых систем приобретать изменения и существовать в различных вариантах называется изменчивостью.
У отдельных клеток и организмов одного вида изменчивость, затрагивая их индивидуальное развитие, проявляется в возникновении отличий между ними. На популяционно-видовом уровне организации жизни это свойство проявляется в наличии генетических различий между отдельными популяциями вида, что лежит в основе образования новых видов. Появление новых видов вносит изменения в межвидовые взаимоотношения в биоценозах. Изменчивость в определенном смысле отражает динамичность организации живых систем и наряду с наследственностью является ведущим фактором эволюции.
98. Механизмы защиты генома от мутагенных воздействий.
Защита осуществляется на нескольких уровнях. Прежде всего, организм старается не допустить попадания химических мутагенов в жизненно важные локусы своего генома. Это достигается двумя путями.
Во-первых, избыточные последовательности нуклеотидов ДНК, экранируя кодирующие последовательности нуклеотидов в геноме эукариот, принимают удар большей части химических мутагенов на себя. Те же цели могут быть достигнуты за счет особой пространственной организации ДНК в конкретных участках генома.
Во-вторых, в клетках имеются многочисленные высоко- и низкомолекулярные ловушки мутагенов, важнейшими из которых являются: маннит, энкефалины, индолы, желчные кислоты и их производные, альфа-токоферол, аскорбиновая кислота, тирозин, серотонин и тд
Обе системы защиты не обладают 100%-й эффективностью. То же можно сказать и о точности функционирования ферментных систем, осуществляющих воспроизведение генетической информации. Поэтому нарушения первичной структуры ДНК неизбежны, но большинство первичных повреждений не превращается в мутации благодаря функционированию систем репарации ДНК.
99. Нетрадиционное наследование (геномный импринтинг, однородительская дисомия).
Сущность геномного импритинга заключается в том, что хромосомы каким-то образом маркируются (или импринтируются) перед слиянием гамет в соответствие со своим родительским происхождением.
В результате ГИ у потомства мутации, унаследованные от матери или от отца, фенотипически проявляются по-разному.
Эффект импринтинга был установлен достаточно определенно для четырех хромосом человека: 7, 11, 14 и 15.
Под геномным импринтингом понимают процесс, который дифференциально маркирует материнские и отцовские гомологичные хромосомы, что приводит к разному фенотипическому проявлению мутаций у потомства, унаследованных от матери или отца. В участках генома, подверженных импринтингу, экспрессируется только один из двух аллелей – отцовский или материнский, т.е. наблюдается моноаллельная экспрессия генов. Второй аллель, вследствие наличия на нем некоего отпечатка, импринтирован (выключен или подавлен) и не экспрессируется. Такой способ регуляции генов свидетельствует о неэквивалентном вкладе родителей в геном потомков.
Геномный импринтинг — эпигенетический процесс, при котором экспрессия определенных генов осуществляется в зависимости оттого, от какого родителя поступил аллель гена. Это ненаследуемый процесс, который не подчиняется наследованию по Менделю. Импринтинг генов вызывает экспрессию аллелей гена полученных от матери в случае генов H19 или CDKN1C и от отца в случае гена IGF2. Импринтинг некоторых генов в составе генома показан для насекомых, млекопитающих и цветковых растений.
Импринтинг генов осуществляется с помощью процесса метилирования ДНК. Если по каким-то причинам импринтинг не сработает, это может привести к появлению генетических нарушений (например Синдром Прадера-Вилли.
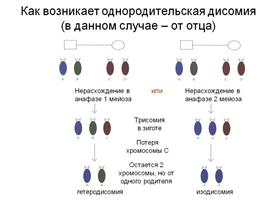
Однородительская дисомия есть наследование обеих копий целой хромосомы или ее части от одного родителя (при отсутствии соответствующего генетического материала от другого родителя).
Существуют 2 типа ОРД:
а) изодисомия, возникающая при нерасхождении хромосом во II делении мейоза, причем обе хромосомы являются копиями и имеют одинаковые последовательности нуклеотидов;
б) гетеродисомия, нерасхождение хромосом в 1 мейотическом делении, с неидентичными гомологами.
На популяционном уровне частота ОРД достаточно высока - порядка 1 на 3000 зигот.
При определении фенотипических последствий ОРД нужно принимать во внимание следующие моменты:
l проявление аутосомно-рецессивного заболевания из-за перехода рецессивных генов в гомозиготное состояние при изодисомии;
l феномен импринтинга.
Однородительская дисомия, то есть наследование обеих копий целой хромосомы или ее части от одного родителя (при отсутствии соответствующего генетического материала от другого родителя), является исключением из менделевских принципов наследования. Она встречается редко и вызывает, например, синдром Прадера-Вилли и синдром Ангельмана .
Роль дисомии в патологии во многом усугубляется геномным импринтингом , который приводит к неодинаковой экспрессии материнской и отцовской копий гена
Возможный механизм дисомии - элиминация лишней хромосомы у плода с трисомией на ранних стадиях эмбриогенеза. Болезнь проявляется в том случае, если элиминируется лишняя хромосома, происходящая из нормальной гаметы
Однородительская дисомия была описана при муковисцидозе , когда оба мутантных аллеля наследовались от одного родителя. В таких случаях дисомия имитирует аутосомно-рецессивное наследование
У 20-30% больных с синдромом Прадера-Вилли , имеющих по данным цитогенетического исследования нормальный кариотип, с помощью
молекулярно-биологических методов обнаруживается дисомия материнской 15-й хромосомы . Отцовская 15-я хромосома у таких больных отсутствует.
Предполагают, что однородительская дисомия является причиной внутриутробной задержки развития , умственной отсталости и микроцефалии . Эти предположения пока не подтверждены молекулярно-биологическими исследованиями.
100. Нетрадиционное наследование (экспансия тринуклеотидных повторов, митохондриальное наследование).
Дата: 2019-07-31, просмотров: 640.