В крови мочевая кислота находится в форме ее солей – уратов натрия. Из-за низкой растворимости ураты способны оседать в зонах с пониженной температурой, например, в мелких суставах стоп и пальцев ног. Накапливающиеся в межклеточном веществе ураты некоторое время фагоцитируются, но фагоциты не способны разрушить пуриновое кольцо. В результате это приводит к гибели самих фагоцитов, к выходу лизосомальных ферментов, активации свободнорадикального окисления и развитию острой воспалительной реакции – развивается подагрический артрит. В 50-75% случаев первым признаком заболевания является мучительная ночная боль в больших пальцах ног. Над участками прикрепления сухожилий в локте, колене или стопе, а также в завитках ушных раковин образуются подагрические тофусы (отложения кристаллов уратов).
Первичная подагра возникает, как правило, у мужчин среднего возраста вследствие избыточного образования и/или снижения экскреции мочевой кислоты. Биохимическая причина в большинстве случаев остается неизвестной, и первичную подагру считают заболеванием с полигенным наследованием. Гиперуриемия и подагра в детском возрасте очень часто бывают вторичными, т.е. обусловленными другими заболеваниями, при которых ускоряется распад тканей и обновление клеток, что и приводит к повышению образования или снижению экскреции мочевой кислоты. Подагра может развиться при любых состояниях, сопровождающихся снижением экскреции мочевой кислоты: при лечении злокачественных новообразований или обезвоживании , при лактатацидозе , кетоацидозе , голодании , применении диуретиков и почечной недостаточности . К повышению уровня мочевой кислоты в сыворотке крови приводит также чрезмерное потребление пуринов , алкоголя или углеводов .
Длительное время подагру считали "болезнью гурманов", однако затем внимание исследователей переместилось к наследственному изменению активности ферментов метаболизма пуринов:
· увеличение активности ФРДФ-синтетазы – приводит к избыточному синтезу пуринов,
· уменьшение активности гипоксантин-гуанин-фосфорибозил-трансферазы – из-за этого ФРДФ не используется для реутилизации пуриновых оснований, а участвует в первой реакции их синтеза. В результате возрастает количество разрушающихся пуринов и одновременно повышается их образование.
Оба ферментативных нарушения рецессивны и сцеплены с X-хромосомой. Подагрой страдает 0,3-1,7% взрослого населения земного шара, соотношение заболевших мужчин и женщин составляет 20 : 1.
Основы лечения
Диета – снижение поступления предшественников мочевой кислоты с пищей и уменьшение ее образования в организме. Для этого из рациона исключаются продукты, содержащих много пуриновых оснований – пиво, кофе, чай, шоколад, мясные продукты, печень, красное вино. Предпочтение отдается вегетарианской диете с количеством чистой воды не менее 2 л в сутки.
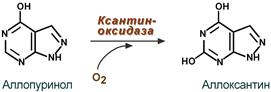 Реакция превращения аллопуринола
Реакция превращения аллопуринола
|
К лекарственным средствам лечения подагры относят аллопуринол, по структуре схожий с гипоксантином. Ксантиноксидаза окисляет аллопуринол в аллоксантин, и последний остается прочно связанным с активным центром фермента и ингибирует его. Фермент осуществляет, образно говоря, самоубийственный катализ. Как следствие, ксантин не превращается в мочевую кислоту, и поскольку гипоксантин и ксантин лучше растворимы в воде, то они более легко выводятся из организма с мочой.
49. При обследовании девочки 3 лет с прогрессирующим отставанием умственного развития в моче определился фенилпируват, а в плазме крови было повышено содержание фенилаланина. Для какой патологии характерны данные признаки?
Фенилкетонурия – группа аутосомнорецессивных заболеваний, обусловленных нарушением обмена незаменимой аминокислоты фенилаланина (ФА), поступающей в организм человека с белковой пищей.
ФКУ объединяет несколько генетически гетерогенных форм нарушения обмена фенилаланина, сходных по клиническим признакам: классическая ФКУ (ФКУ I типа), обусловленная дефицитом фенилаланин-4- гидроксилазы (ФАГ) и птерин-зависимые формы гиперфенилаланинемии, связанные с дефектом птеринового кофактора (ФКУ II и III типов).
Классическая фенилкетонурия (фенилкетонурия I типа) обусловлена дефицитом фермента фенилаланингидроксилаза (ФАГ), ведущим к накоплению фенилаланина и продуктов его распада в биологических жидкостях.
Птерин-зависимые формы ФКУ (2 и 3 тип) имеют сходные с классической ФКУ клинические проявления. При этих формах основную роль в патогенезе играет резкая недостаточность нейромедиаторов катехоламинового и серотонинового ряда, что делает изолированную диетотерапию бесперспективной и требует иного принципа лечения. В комплекс лечения таких больных должны входить ВН4 или его синтетические аналоги.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Классическая фенилкетонурия:
Первыми проявлениями болезни служат вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги, признаки аллергического дерматита. Отчетливо формируется задержка статикомоторного и психоречевого развития, возможно формирование микроцефалии и гидроцефалии. Характерны такие фенотипические особенности как гипопигментация кожи, волос, радужной оболочки глаз. Обращает внимание своеобразный «мышиный» запах мочи больных.
Эпилептические приступы занимают важное место в клинической картине фенилкетонурии. Они встречаются почти у половины больных и в некоторых случаях могут служить первым признаком болезни
Патогенез
В результате метаболического блока превращения фенилаланина в тирозин происходит значительное накопление фенилаланина и его токсических метаболитов (фенилпировиноградной, фенилмолочной, фенилуксусной кислот, фенилэтиламина и др.) в биологических жидкостях больного организма, что оказывает токсическое действие на центральную нервную систему.
Несмотря на многие научные исследования, патогенез дисфункции головного мозга до сих пор не полностью выяснен. В последнее время все большее значение в патогенезе ФКУ придается нарушениям обмена моноаминовых нейромедиаторов (катехоламинов и серотонина), играющих исключительно важную роль в созревании и функционировании центральной нервной системы.
Диагностика
В родильном доме у всех новорожденных на 4-й день жизни (у недоношенных на 7-й день) берется кровь из пятки на тест-бланки (приложение 1), которые доставляются в лабораторию медико-генетической консультации, осуществляющей определение содержания ФА в крови.
Используют микробиологический и флюорометрический методы определения концентрации фенилаланина в крови, а также пробу Фелинга на фенилпировиноградную кислоту в моче (прибавление нескольких капель 5% раствора треххлористого железа и уксусной кислоты к моче больного приводит к появлению зеленой окраски пятна на пеленке). Эти и другие подобные методы относятся к категории ориентировочных, поэтому при положительных результатах требуется специальное обследование с использованием точных количественных методов определения содержания фенилаланина в крови и моче (хроматография аминокислот, использование аминоанализаторов и др.), которое осуществляется централизованными биохимическими лабораториями.
Значение фенилаланина выше 2,0 мг/дл классифицируется как ГФА, которая требует проведения уточняющей диагностики. Это более сложная процедура, иногда многоэтапная. Во-первых, необходимо подтвердить гиперфенилаланинемию и, во-вторых, установить ее причину. ГФА может быть обусловлена классической (типичной) ФКУ, связанной с недостаточностью фенилаланингидроксилазы, птерин-зависимыми (кофакторными) формами болезни, резистентными к диетотерапии, наследственной гиперфенилаланинемией (доброкачественной ГФА), другими формами нарушения метаболизма (тирозинемия, галактоземия и др.).
Следующим этапом для уточнения классической фенилкетонурии является молекулярно-генетическая диагностика. В большинстве лабораторий существуют наборы, позволяющие определять частые мутации в гене ФАГ, имеющиеся у 80% больных ФКУ. При отсутствии исследуемых мутаций у пациента рекомендуется проведение секвенирования гена РАН. Для исключения птерин-зависимых форм ФКУ во многих развитых странах у лиц с гиперфенилаланинемией исследуются птерины в моче.
На заключительном этапе проводится медико-генетическое консультирование семьи, планируется пренатальная диагностика
ЛЕЧЕНИЕ
Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ; ее основной целью является предупреждение развития повреждения ЦНС, нарушений физического и интеллектуального развития. Диетотерапия должна быть начата не позднее первых недель жизни ребенка. Она основана на резком ограничении фенилаланина в рационе больных детей за счет исключения высокобелковых продуктов. Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина.
При организации диетотерапии необходимо учитывать:
- клиническую форму заболевания;
- уровень ФА в крови;
- возраст ребенка;
- нутритивный статус (физическое развитие);
- толерантность ребенка к пищевому ФА;
- количество ФА и натурального белка, получаемого с пищей;
- количество основных пищевых веществ и энергии в лечебном рационе.
В большинстве стран натуральные продукты, на которых строится рацион, по содержанию в них белка делят на три группы, составляющие так называемый «пищевой светофор» больного ФКУ
· В красный список входят продукты с высоким содержанием ФА, которые полностью исключаются из рациона больных ФКУ.
· Желтый список – продукты, которые содержат умеренное количество белка и, следовательно, фенилаланина, и поэтому должны использоваться с осторожностью, в небольших количествах и под систематическим контролем ФА крови. Они также должны равномерно распределяться в течение дня.
· Зеленый список – продукты, содержащие незначительное количество фенилаланина, которые могут применяться совершенно свободно в обычных количествах
Дата: 2019-05-28, просмотров: 856.