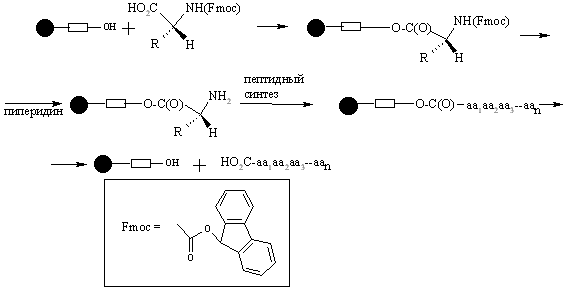
История медицинской химии
Впервые курс медицинской химии был прочитан в МГУ в 1998 году. Как наука медицинская химия сформировалась к 1970 году.
В 19 веке медицинская химия начала прогрессировать. Были выделены средство против малярии из хинного дерева, морфин, салициловая кислота (противомикробное средство).
В 1888 году Байер впервые в мире поставил промышленное производство фенацетина
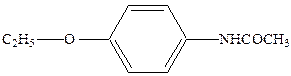
а в 1899 году – производство аспирина
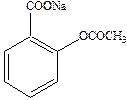
К этому времени люди научились анализировать белки, жиры и углеводы, которые являются основными мишенями действия лекарственных препаратов.
В 1910 году Пауль Эрлих изобрел средство от сифилиса – сальварсан
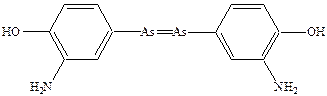
Это обусловило возникновение концепции химиотерапии. Концепция химиотерапии предполагает не просто применение химических веществ для лечения патологий, но и необходимость модификации структур предполагаемых лекарственных соединений с тем чтобы максимально эффективно воздействовать на пораженный орган.
Эрлих также разработал теорию рецепторов и структурных изменений физиологически активных органических соединений, происходящих при их взаимодействии с рецепторами. Эта теория явилась отправной точкой для медицинской химии.
В 1928 году был открыт пенициллин, а в 1944 году Ваксберг открыл стрептомицин. Так началась эра антибиотиков.
В 60-ые годы были разработаны психотропные препараты: аминазин, мекробамат (транквилизатор).
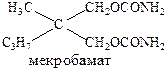
а также резерпин, метилдофа (сердечно-сосудистые препараты).
Все эти препараты были открыты «на ощупь». Ученые продолжали искать ФАВ.
ФАВ (БАВ) – физиологически (биологически) активные вещества.
С другой стороны, проникновение компьютерных методов в органическую химию привело к бурному развитию методов расчета структуры молекул (геометрия и конформация, заряды, молекулярные орбитали, электростатические потенциалы, топологические индексы и т.д.). В силу этого количественное описание структурных особенностей даже очень сложных молекул биологического уровня становится обычным инструментом химика-органика.
В 70-ых годах была создана методологическая основа для возникновения и использования рациональных подходов к синтезу ФАВ, которая называется drug design. Это привело к формированию медицинской химии с ее современным аппаратом.
Медицинская химия – междисциплинарная наука, в ней задействованы органическая, биоорганическая, фармацевтическая химии, фармакология и биохимия. Но они не дают ответа на вопросы: «Какую структуру надо синтезировать, чтобы создать ФАВ?», «Какую структурную формулу спрогнозировать для конкретного кандидата в лекарство?». Эти вопросы являются центральными для медицинской химии.
Предметом медицинской химии является поиск и структурный дизайн ФАВ, выявление взаимосвязи химической структуры и физиологической активности и решение обратной задачи «строение-свойство», а именно конструирование необходимых структур обладающих заданным свойством.
Процесс поиска и конструирования включает в себя три основные стадии:
- Поиск соединения лидера (lead compound).
- Оптимизация соединения лидера.
- Разработка лекарственного препарата.
Стратегия поиска лекарственных препаратов зависит от накопленных знаний о препаратах, мишени, их взаимодействия и т.д. Под названием «мишень» понимают клетки, ткани, органы, функциональные системы, ферменты, акцепторы, рецепторы.
Рецепторы по Эрлиху – это небольшой участок химически определенный на большой молекуле протоплазмы, участвующей в питании и метаболизме клетки, и способный связывать специфические антигены или лекарственные вещества.
Антигены – вещества, которые воспринимаются организмом как чужеродные и вызывают специфический иммунный эффект.
Рецепторы – главным образом белковые структуры, функции которых заключаются в узнавании химического сигнала и последующей его трансформации в адекватный ответ клетки. Другими словами рецепторы представляют собой материальные субстраты активности и чувствительности клеток.
Возможны четыре ситуации при начале поиска лекарственного средства:
- Структура как рецептора, так и лиганда неизвестны. Рецептор является мишенью лекарственного вещества в организме, а понятие лиганд предполагает любое эндогенное (endon – внутри) соединение, взаимодействующее с этим рецептором в организме.
- Известна только структура рецептора.
- Известна только структура лиганда.
- Известна структура как рецептора, так и лиганда.
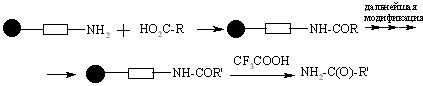
Комбинаторный синтез
В недавнем прошлом открытие лекарственного средства начиналось с индивидуального синтеза сотен, а иногда и тысяч аналогов малоактивного препарата, в надежде повысить его активность и селективность, с одновременным снижением токсичности. В среднем, для достижения поставленной цели необходимо было синтезировать более 10 000 соединений, а чтобы получить это молекулярное множество, один человек должен был бы работать около 1000 лет.
В настоящее время поиск новых лекарственных препаратов строится на принципиально новой основе.
Теперь все начинается с расшифровки структуры генов, кодирующих белки, ответственные за проявление тех или иных биологических функций. Затем эти “мишени” (targets) получают в чистом виде и создают специальные тестовые системы, позволяющие уже не in vivo, а микрометодами, in vitro, определять биологическую активность как давно синтезированных так и вновь получаемых веществ.
Таким образом находят биологически активную структуру (hit compounds), которую модифицируя преобразуют в соединение-лидер (lead compounds), в результате оптимизации свойств которого получают лекарственное соединение (clinical candidate). Схематично этот процесс можно выразить следующим образом:
Идентификация мишени
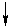
Синтез молекулярного множества, скрининг
Определение строения и идентификация хита
Оптимизация хита
Создание соединения-лидера
Оптимизация соединения-лидера
Потенциальное лекарственное средство
Успехи геномики свидетельствуют о том, что в ближайшее десятилетие будет получено около 2000 новых мишеней и темп скрининга придется повышать, несмотря на то, что уже в настоящее время удается подвергнуть скринингу до 100 тысяч соединений в расчете на одну мишень. Поэтому если ранее скрининг отставал от синтеза, то теперь ситуация изменилась: вдобавок к образцам сотен тысяч уже известных органических веществ, составляющих так называемые исторические библиотеки, понадобилось быстро получать миллионы соединений.
Подобную задачу способно решить только особое направление в органической химии, так называемый комбинаторный синтез, позволяющий специальными приемами быстро синтезировать обширные коллекции веществ с похожей структурой, так называемые библиотеки.
Комбинаторный синтез – это одна из наиболее быстро развивающихся в последнее время областей фармацевтической индустрии. Он является наиболее важным инструментом для создания новых лекарственных препаратов. Что же такое комбинаторный синтез и почему он так важен?
Говоря попросту, комбинаторный синтез является способом получения большого числа соединений за короткое время. При этом используются обычные реакционные пути и традиционный набор исходных материалов и реагентов.
Обычно химик получает соединение, которое он выделяет, очищает и идентифицирует. При комбинаторном синтезе делается акцент на получение смеси. Строение соединения необязательно устанавливать. Необязательно выделять и очищать. Вместо этого каждую смесь тестируют на биологическую активность целиком. Если смесь не проявляет активность, то нет необходимости эту смесь более изучать и ее сохраняют. Если активность проявляется, то уже требуется идентификация каждого компонента смеси. В известном смысле комбинаторный синтез выглядит как синтетический эквивалент природного первичного океана-бульона, в котором происходил в течение эволюции случайный синтез различных химических молекул, немногие из которых проявляли биологическую активность.
Комбинаторный синтез используется в основном в двух направлениях:
Жидкофазный синтез
Преимущества:
- Жидкофазный синтез возможен при использовании всех известных синтетических методов без каких либо ограничений
- Реакция проходит в гомогенных условиях
- Можно легко использовать нагревание
- Реакцию можно контролировать
- Возможна очистка и анализ продуктов реакции на каждой стадии
Недостатки:
- После окончания реакции все целевые соединения и побочные продукты находятся в смеси и требуется их разделение
- При использовании избытка реагентов, для достижения хороших выходов продуктов, эти реагенты необходимо тщательно очищать
- В том случае, если реагенты, продукты и побочные соединения невозможно перегнать или они не выпадают в осадок, их можно разделить или очистить только экстракцией или хроматографически, что обычно требует большого времени
- Автоматизация процессов очистки соединений в растворе весьма затруднительна
Процессы создания библиотек путем реакций в растворе делятся на параллельный синтез и синтез смесей.
Параллельный синтез протекает как обычные реакции, только их много, и они идут одновременно в отдельных реакционных сосудах. В этом случае в конце превращения регенты и побочные продукты летучи, их выпаривают. Если это невозможно, иногда используют кислотно-основную экстракцию или колоночную хроматографию. Это обычно затруднительно при наличии большого числа получаемых соединений.
Все это приводит к тому, что жидкофазный способ создания библиотек относительно мало распространен.
Твердофазный синтез
В органической химии нет ни одной реакции, обеспечивающей на практике количественные выходы целевых продуктов в любом случае. Единственное исключение составляет, по-видимому, полное сжигание органических веществ в кислороде при высокой температуре до СО2 и Н2О. Поэтому очистка целевого продукта является сложной и трудоемкой задачей. Например, 100%-ная очистка продуктов пептидного синтеза является трудноразрешимой проблемой. Действительно, первый полный синтез пептида, гормона окситоцина (1953 г), содержащего всего 8 аминокислотных остатков, рассматривался как выдающееся достижение, принесшее его автору, В. дю Виньо, Нобелевскую премию 1955 г. Однако уже в следующие двадцать лет синтезы полипептидов подобной сложности превратились в рутину, так что в настоящее время синтез полипептидов, состоящих из 100 и более аминокислотных остатков, уже не рассматривается как непреодолимо трудная задача. Что вызвало столь драматические изменения в области синтеза полипептидов?
Дело в том, что в начале 60-х годов был предложен новый подход к решению проблем выделения и очистки, возникающих в пептидном синтезе. Позже автор открытия этого подхода, Р.Б. Меррифилд, в своей Нобелевской лекции рассказал, как это произошло: “Однажды у меня возникла мысль о том, как может быть достигнута цель более эффективного синтеза пептидов. План состоял в том, чтобы собирать пептидную цепь постадийно, причем во время синтеза цепь должна быть одним концом привязана к твердому носителю”. В результате выделение и очистка промежуточных и целевых производных пептидов сводились просто к фильтрованию и тщательной промывке твердого полимера для удаления всех избыточных реагентов и побочных продуктов, остающихся в растворе. Такая механическая операция может быть выполнена количественно, легко стандартизируется и может быть даже автоматизирована. Рассмотрим эту процедуру более подробно.
Полимерный носитель в методе Меррифилда – это гранулированный сшитый полистирол, содержащий хлорметильные группы в бензольных ядрах. Эти группы превращают полимер в функциональный аналог бензилхлорида и сообщают ему способность легко образовывать сложноэфирные связи при реакции с карбоксилат-анионами. Конденсация такой смолы с N-защищенными аминокислотами ведет к образованию соответствующих бензиловых эфиров. Удаление N-защиты из дает С-защищенное производное первой аминокислоты, ковалентно связанное с полимером. Аминоацилирование освобожденной аминогруппы N-защищенным производным второй аминокислоты с последующим удалением N-защиты приводит к аналогичному производному дипептида также привязанному к полимеру:
Такой двустадийный цикл (удаление защиты-аминоацилирование) может быть, в принципе, повторен столько раз, сколько требуется для наращивания полипептидной цепи заданной длины.
Использование твердого носителя само по себе еще не может упростить решение проблемы отделения n-звенного пептида от его (n-1)-членного предшественника, поскольку оба они привязаны к полимеру. Однако этот подход позволяет безопасно использовать большие избытки любого реагента, необходимые для достижения практически 100%-ной конверсии (n-1)-членного предшественника в n-членный пептид, так как привязанные к носителю целевые продукты на каждой стадии могут быть легко и количественно освобождены от избыточных реагентов (что было бы весьма проблематично при работе в гомогенных системах).
Сразу же стало понятно, что возможность очистки продукта после каждой реакции путем простого фильтрования и промывки, и то, что все реакции можно проводить в одном реакционном сосуде, составляют идеальные предпосылки для механизации и автоматизации процесса. Действительно, всего три года потребовалось для разработки автоматической процедуры и аппаратуры, позволяющих выполнять программируемый синтез полипептидов с заданной последовательностью аминокислотных остатков. Первоначально и сама аппаратура (емкости, реакционные сосуды, шланги), и система управления были очень примитивны. Тем не менее, мощь и эффективность общей стратегии были убедительно продемонстрированы рядом пептидных синтезов, выполненных на этом оборудовании. Так, например, с помощью такой полуавтоматической процедуры был успешно выполнен синтез природного гормона инсулина, построенного из двух полипептидных цепей (состоящих из 30 и 21 аминокислотных остатков), связанных дисульфидным мостиком.
Твердофазная техника приводила к существенной экономии труда и времени, необходимых для пептидного синтеза. Так, например, ценой значительных усилий Хиршмен с 22 сотрудниками завершили выдающийся синтез фермента рибонуклеазы (124 аминокислотных остатка) с помощью традиционных жидкофазных методов. Почти одновременно тот же белок был получен путем автоматизированного твердофазного синтеза. Во втором случае синтез, включающий 369 химических реакций и 11 931 операцию, был выполнен двумя участниками (Гатте и Меррифилд) всего за несколько месяцев (в среднем до шести аминокислотных остатков в день присоединялись к растущей полипептидной цепи). Последующие усовершенствования позволили построить полностью автоматический синтезатор.
Метод Меррифильда и послужил основой для нового направления органического синтеза – комбинаторной химии.
Хотя иногда комбинаторные эксперименты проводятся в растворах, но в основном, они осуществляются с использованием твердофазной техники – реакции протекают с использованием твердых подложек в виде сферических гранул полимерных смол. Это дает ряд преимуществ:
- Различные исходные соединения могут быть связаны с отдельными гранулами. Затем эти гранулы смешиваются и, таким образом, все исходные соединения могут взаимодействовать с реагентом в одном эксперименте. В результате продукты реакции образуются на отдельных гранулах. В большинстве случаев, смешивание исходных в традиционной жидкой химии приводит обычно к неудачам – полимеризации или осмолению продуктов. Эксперименты на твердой подложке исключают эти эффекты.
- Поскольку исходные материалы и продукты связаны с твердой подложной, то избыток реагентов и не связанных с подложкой продуктов можно легко отмыть от полимерной твердой подложки.
- Можно использовать большие избытки реагентов, для того чтобы провести реакцию до конца (больше, чем 99%), поскольку эти избытки легко отделяются.
- В случае использования низких объемов загрузок (менее 0,8 ммоль на грамм подложки) можно исключить нежелательные побочные реакции.
- Интермедиаты в реакционной смеси связаны с гранулами и их нет необходимости очищать.
- Индивидуальные гранулы полимера могут быть разделены в конце эксперимента и таким образом получаются индивидуальные продукты.
- Полимерная подложка может быть регенерирована в тех случаях, когда подобраны условия разрыва и выбраны соответствующие якорные группы – линкеры.
- Возможна автоматизация твердофазного синтеза.
Необходимыми условиями проведения твердофазного синтеза, кроме наличия нерастворимой полимерной подложки, инертной в реакционных условиях, являются:
- Присутствие якоря или линкера – химической функции, обеспечивающей связь подложки с наносимым соединением. Он должен быть ковалентно связан со смолой. Якорь также должен являться реакционно-способной функциональной группой для того, чтобы субстраты могли взаимодействовать с ним.
- Связь, образующаяся между субстратом и линкером должна быть стабильна в условиях реакции.
- Должны существовать способы разрыва связи продукта или интермедиата с линкером.
Рассмотрим подробнее отдельные компоненты твердофазного метода синтеза.
Твердая подложка
Как сказано выше, первыми типами смол, которые использовал Меррифильд, были полистирольные гранулы, где стирол был сшит с 1% дивинилбензола. Гранулы были модифицированы хлорметильными группами (линкер), с которыми аминокислоты могли быть соединены через эфирные группы. Эти эфирные связи стабильны в реакционных условиях, которые применялись для петидного синтеза.
Одним из недостатком полистирольных гранул является то факт, что они гидрофобны, тогда как растущая пептидная цепь гидрофильна. В результате, иногда растущая пептидая цепь не сольватируется и сворачивается за счет образования внутримолекулярных водородных связей. Такая форма затрудняет доступ новых аминокислот к концу растущей цепи. Поэтому часто используются более полярные твердые подложки, такие как полиамидные смолы. Такие смолы более пригодны для непептидного комбинаторного синтеза.
Выбор твердой подложки
Синтетические подходы к получению библиотек часто определяются природой выбранной полимерной подложки. Гранулированный полимер должен соответствовать некоторым критериям, в зависимости от стратегий синтеза и скрининга.
Для получаемых библиотек имеют важное значение размер и однородность гранул, а также устойчивость смолы к формированию кластеров. Способность смолы к набуханию в органической и водной среде особенно важна, когда используются обязательные пробы для скрининга структуры, находящейся еще на грануле.
Основные типы полимерных смол для комбинаторного синтеза используемые в настоящее время:
- Полистирол, сшитый с 0,5-2% дивинилбензола (StratoSpheres)
- Полиэтиленгликоль, привитый на сшитом сополимере полистирол- 1% дивинилбензол (TentaGel, AgroGel, NovaGel)
- Полиэтиленгликоль, привитый на 1% сшитый полистирол (PEG-PS)
- Полистирольная макропористая смола с высокой степью сшивки (AgroPore, TentaPore)
- Сополимер бис-2-акриламидполиэтиленгликоль-моноакриламидо-полиэтиленгликоль (PEGA)
- Диметилакриламид нанесенный на макропористую матрицу кизельгура (Pepsyn K)
- Диметилакриламид нанесенный на макропористую матрицу – сшитый 50% полистирол-дивинилбензол (Polyhipe)
Хотя классические гранулированные смолы больше подходят для комбинаторного синтеза библиотек соединений, иногда используются альтернативные носители.
Например, целлюлоза является хорошей подложкой для многократного “капельного синтеза” пептидов или для синтеза библиотек на бумаге. “Капельные” синтезы проводят путем капания растворов защищенных аминокислот на модифицированную бумагу в присутствии активирующего реагента. Здесь реакционным сосудом является непосредственно носитель и нет необходимости манипуляций, характерных для жидких сред в течение синтеза (обычно встряхивание в случае твердофазного синтеза). Реакция идет за счет диффузии жидкости в носителе. Этот принцип внутреннего объемного синтеза был проверен при использовании полимерных носителей на синтезаторе, использующем центрифугирование для устранения жидкости. Было найдено, что капельная техника сопоставима с классическим функционированием твердой фазы в пептидном синтезе.
Было также найдено, что хлопок (вата), как самая чистая форма целлюлозы может служить удобной подложкой твердой фазы, особенно для множественного синтеза или генерирования библиотеки.
Хотя гранулы и являются наиболее распространенной формой твердой положки, но и другие виды (например, иглы) могут также использоваться для комбинаторного синтеза. Модифицированная стеклянная поверхность также может быть применена для олигонуклеотидного синтеза.
Линкеры
Линкер – это молекулярный фрагмент, ковалентно связанный с твердой подложкой. Он содержит реакционноспособные функциональные группы, с которыми взаимодействует первый реагент и который в результате становится связанным со смолой. Образующаяся связь должна быть стабильной в реакционных условиях, но легко разрываться на конечной стадии синтеза.
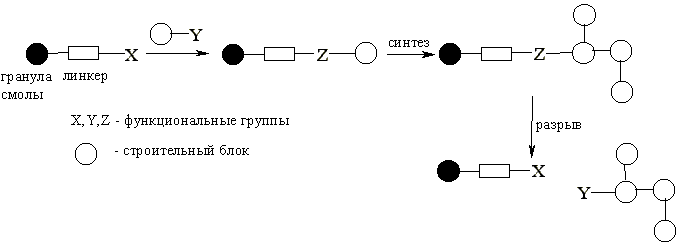
Различные линкеры используются в зависимости от того, какая функциональная группа присутствует в субстрате и от того, какая функциональная группа должна быть сформирована в конце процедуры.
В практике комбинаторного синтеза чаще всего используются следующие линкеры:
- Хлорметильный (-CH2Cl),
- Гидроксильный (-OH),
- Аминный (-NH2),
- Альдегидный (-CHO),
- Силильный (-OSiR3).
| Тип линкера | Тип смолы | Что присоединяет | Что синтезирует | Чем осуществляется разрыв |
| Галогенметил | 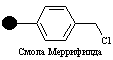
| Карбоновые кислоты, спирты, фенолы, тиолы, амины | Кислоты, спирты, сложные эфиры, тиоэфиры | TFMSA, H2/Pd, i-Bu2AlH, MeONa, HF |
| Галогенметил | 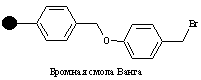
| Алкил и ариламины | Анилиды и сульфамиды | CF3COOH, SOCl2/CF3COOH |
| Галогенметил | 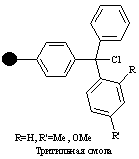
| Спирты, кислоты, фенолы, тиолы, амины | Спирты, кислоты, тиолы, амины, сложные эфиры | 1-5% CF3COOH, 30% гексафторизопропанол |
| Гидроксил | 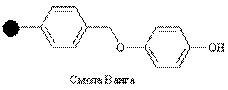
| Спирты, кислоты | Спирты, кислоты, амиды | CF3COOH, амин/AlCl3, i-Bu2AlH |
| Гидроксил | 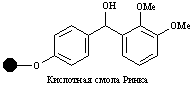
| Спирты, кислоты | Спирты, кислоты | 5% CF3COOH, 10% AcOH |
| Гидроксил | 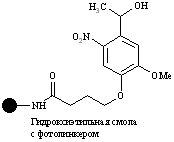
| Кислоты | Кислоты | Свет с длиной волны 365 нм. Линкер стабилен к CF3COOH и пиперидину |
| Гидроксил | 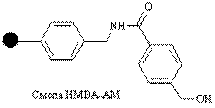
| Кислоты | Амиды кислот, спирты, сложные эфиры, гидразиды | Нуклеофилы (NaOH,NH3/MeOH, NaBH4/EtOH, MeOH/CF3COOH, NH2NH2/DMF |
| Гидроксил | 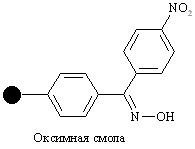
| Защищенные пептиды, ки-слоты | Циклические пепти-ды, мочевины | 25% CF3COOH, гидразиды |
| Гидроксил Линкер Ринкера | 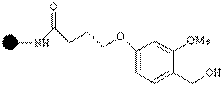
| Спирты, кисло-ты, фенолы | Спирты, кислоты, фенолы | 1-5% CF3COOH |
| Амино | 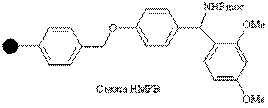
| Кислоты | Карбоксамиды | 95% CF3COOH |
| Амино | 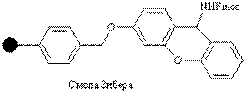
| Кислоты | Защищенные амиды | 1% CF3COOH |
| Амино | 
| Кислоты | Альдегиды и кетоны | LiAlH4 и реактивы Гриньяра |
| Амино | 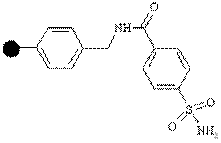
| Карбоновые кислоты | Амиды или карбоновые кислоты | Активация сульфонамида диазометаном или бромацетонитрилом с последующей атакой нуклеофилом амина или гидроксида |
| Альдегид | 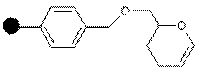
| Первичные или вторичные спирты | Спирты | 95% CF3COOH/H2O или CF3COOH/CH2Cl2/EtOH |
| Альдегид | 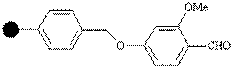
| Амины | Карбоксамиды, сульфонамиды | CF3COOH |
Смолы Ванга могут быть использованы в пептидном синтезе посредством N-защищенной аминокислоты, связанной с линкером эфирной связью. Такая эфирная связь устойчива к сочетанию и стадии снятия защиты, но может быть разрушена трифторуксусной кислотой для снятия конечного пептида с гранулы смолы.
Субстраты с карбоксильной группой могут быть связаны со смолой Ринка через амидную связь. Как только процедура заканчивается, взаимодействие с трифторуксусной кислотой освобождает продукт с первичной амидной группой.
Первичные и вторичные спирты могут быть связаны со смолой, модифицированной дигидропираном. Связывание спирта происходит в присутствии 4-толуолсульфоната в дихлорметане. Снятие продукта происходит с использованием трифторуксусной кислоты:
Основные принципы
Комбинаторный синтез часто используется для получения смесей продуктов. Для этого используется широкий набор исходных материалов и реагентов. Это не значит, что все возможные исходные материалы сразу помещены один реакционный сосуд. Планирование комбинаторного синтеза означает стремление к минимизации труда и максимизации числа получаемых структур.
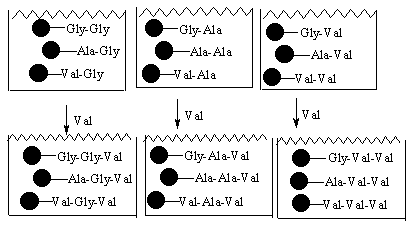
Для примера, предположим, что мы хотим синтезировать все возможные дипептиды из пяти различных аминокислот. Используя традиционную химию их можно получать по одному. Таким образом, 25 возможных дипептидов будут получены в 25 отдельных экспериментах.
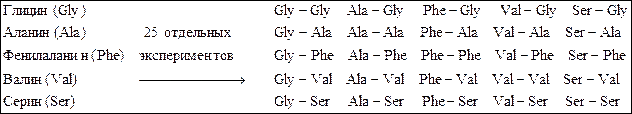
Однако используя комбинаторную химию все они могут быть получены с гораздо меньшим трудом. Если все пять различных аминокислот отдельно связать с гранулами смолы, то эти гранулы затем могут быть смешаны вместе. Таким образом, можно получить все возможные дипептиды путем взаимодействия со второй аминокислотой за пять экспериментов. Например, в одном эксперименте пять различных аминокислот будут реагировать с глицином, давая пять из 25 возможных дипептидов:
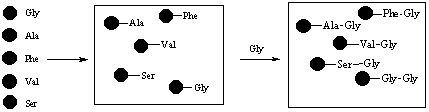
Эта смесь затем может быть протестирована на биологическую активность. Если результаты положительные, то усилия будут направлены на идентификацию того дипептида, который проявляет активность. Если активности нет, то эта смесь будет отставлена и отправлена на хранение. Полученные смеси не выбрасываются. Хотя они и не содержат соединения-лидера, но они могут содержать лидер для другой области медицинской химии. Все эти смеси как активных, так и не активных соединений, образующихся при комбинаторном синтезе, сохраняются и носят название “комбинаторных библиотек”. Последовательная обработка связанных аминокислот другими кислотами из приведенного набора даст 5 смесей состоящих из 25 пептидов.
Мы привели пример получения 25 соединений за пять синтезов. Однако, используя комбинаторный синтез имеется возможность получения тысяч или даже миллионов соединений.
Принцип “смешай и раздели”
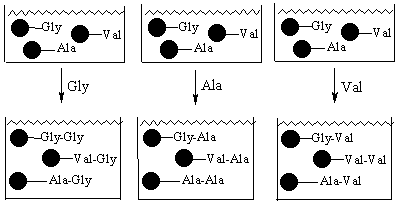
В случае когда генерируются большие количества различных структур очень важно минимизировать объем работ. В этом случае очень хорошие результаты дает так называемый принцип смешивания и разделения (mix and split method). Этот принцип лучше проиллюстрировать на примере. Предположим, что мы хотим получить все возможные трипептиды из трех различных аминокислот (например, Gly, Val, Ala). Для это применим метод смешивания и разделения:
Стадия 1 – свяжем каждую аминокислоту с твердой подложкой
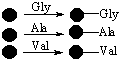
Стадия 2 – смешаем все гранулы вместе и поделим смесь на три равные части
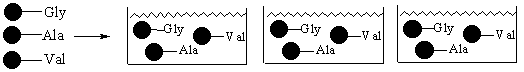
Стадия 3 – проведем реакции каждой части с разными аминокислотами
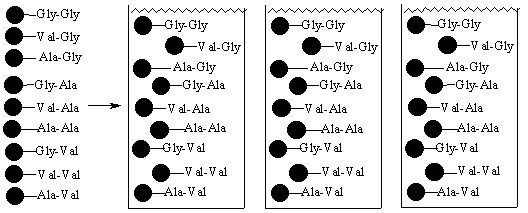
Стадия 4 – выделим все гранулы, смешаем затем их все вместе и поделим
на три равные части. Каждая часть сейчас содержит все 9 возможных дипептидов
Стадия 5 – проведем реакции каждой порции с тремя аминокислотами
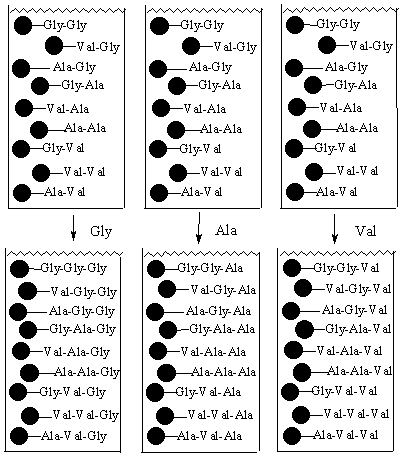
В результате мы получили все 27 возможных трипептида всего за три эксперимента вместо 27 по классической схеме. В этом примере происходило связывание между собой аминокислот, но по эту стратегию можно использовать для связывания вместе и других мономерных единиц или комбинации химических структур.
Этот принцип используется в методе комбинаторного синтеза “одна гранула – одно соединение” (One-Bead – One-Compound или ОГОС). В этом случае одна гранула связывается только с одим типом молекул, хотя их может находиться на этой грануле диаметром 100 микрон до триллиона копий. Этот метод позволяет, с одной стороны, создавать библиотеки олигомеров. Описаны созданные на основе этого метода библиотеки пептидов из 20 природных L-аминокислот, олигонуклеотидов, олигосахаридов, пептоидов, олигокарбаматов, олигомочевин и др.
Другое очень важное направление применения метода ОГОС – получение библиотек небольших молекул. Эти методом созданы библиотеки бензодиазепинов, гидантоинов, кубанов, лактамов, тиазолидинов, тетрагидрофуранов, бензпиранов, изоксазолов, триазолов, циклопентанов, циклогексанов, пиперазинов, дикетопиперазинов, пиридинов, хинолинов, бенимидазолов, хиназолинов, триазинов и др.
Смесь реагентов
Второй синтетический подход, названный методом “смесь реагентов”, предполагает использование избытков реагентов в отношении каждой реакционного цента на твердофазном субстрате. Применение таких смесей реагентов требует полного знания механизма и кинетики процессов, используемых в выполняемых реакциях. Большой избыток поступающих реагентов используется для того, чтобы наблюдалась реакция кинетики псевдопервого порядка. Однако этот подход имеет существенные ограничения. Очень важно, чтобы относительные скорости реакций вводящихся реактивов были приблизительно равными, т.е эти реагенты должны иметь примерно равную нуклеофильность и отсутствие стерических помех. Такие требования довольно трудно выполнять. Тем не менее было обнаружено, что эта концепция хорошо применима к синтезу гетероциклических библиотек типа циклической мочевины и тиомочевины.
Фотолитография
Фотолитография – это техника для миниатюризации комбинаторного синтеза. В синтезе пептидов поверхность твердой подложки состоит из аминогрупп, защищенных фотолабильной нитровератрилоксикарбонильной защитной группой (NVOC). Используя маску – защищая часть поверхности от облучения светом, удается снять защиту в экспонированной области.
После этого плата обрабатывается аминокислотой и в результате реакция протекает только в той области на плате, где предварительно была снята защита. Плата затем промывается для удаления избытка аминокислоты. Затем процесс может повторен в другой области платы, используя другую маску и таким образом различные пептидные цепи могут быть построены на различных частях платы, последовательность которых известна по записям на используемых масках.
Затем проводится инкубация платы с белком рецептора, для определения активного соединения, которое закреплено в соответствующем положении платы. Удобный метод для осуществления этого процесса – это использование платы с флуресцентно-меченым рецептором. В этом случае только в присутствия активного соединения будет наблюдаться флуоресценция. Интенсивность флуоресценции может быть измерена методом флуоресцентной микроскопии и, таким образом, можно определить сродство синтезированного соединения к рецептору. Альтернативным способом может быть использование радионуклидов или хемолюминисценции.
Возможности этого метода очень высоки. При 20-микронном разрешении может быть приготовлена плата, на которой находится 250 000 различных соединений на квадратном сантиметре.
В случае пептидного синтеза общие операции, такие как снятие защиты и промывание, производятся путем погружения платы в большие бани, но связывание производится в ячейках, таким образом, чтобы каждая ячейка содержала уникальную аминокислоту.
Добавление реагента и удаление его избытка, а также нагревание и охлаждение реакционных смесей, может осуществляться автоматически. Такие синтезаторы подходят как для множественного параллельного синтеза индивидуальных соединений, так и для параллельного синтеза смесей, содержащих различные, но структурно подобные соединения.
Микроманипуляция
Каждый гранула в смеси содержит только один тип продукта. Следовательно, индивидуальная гранула может быть отделена, продукт очищен и затем протестирован. Эту процедуру можно осуществить методом калориметрического анализа, которым продукты тестируются на активность. Активные гранулы отличаются по цветным реакциям и могут быть выбраны путем микроманипуляции (ручным способом).
Обратная развертка
Микроманипуляция имеет серьезные трудности и возможности ошибок, в том случае когда имеет дело с большими количествами гранул. Для уменьшения объема работ в этом случае можно применить метод, известный как обратная развертка. Его можно проиллюстрировать на примере библиотеки трипептидов, которая описана ранее. В этом примере было синтезировано три смеси. Было предположено, что одна из них имеет активный компонент. Как найти, какой из девяти возможных трипептидов обладает активностью?
Мы можем синтезировать отдельно все девять пептидов и проверить на активность каждый из них. Однако, можно сократить объем работы, если сохранить образцы димерных смесей, полученных в процессе комбинаторного синтеза.
Предположим, что третья смесь трипептидов, описанных ранее, показывает активность. Это значит, что активный трипептид имеет валин на конце цепи. На следующей стадии возьмем три дипептидных смеси, которые мы сохранили и проведем связывание каждой из их с валином. В этом случае мы получим девять трипептидов разделенных на три отдельных смеси.
Теперь нам известны вторая и третья аминокислота в каждой смеси. Все три смеси снова протестируем на биологическую активность. Если одна из них проявляет активность, мы можем знать последовательность в ней второй и третьей аминокислоты. Предположим, что смесь содержащая Ala и Val является активной смесью. Тогда мы можем синтезировать индивидуально уже не девять, а только три трипептида, находящиеся в этой смеси и, проведя тест, определить активное соединение.
Применение метода смешивания и разделения вместе с методом обратной развертки дают значительную экономию объема работ.
Последовательное разделение
Линкеры являются устройством, которое позволяет отделить определенную долю продукта от гранулы. Если смесь активна, то она может быть разделена на меньшие по составу смеси и конечный продукт может быть отделен от гранул и протестирован.
Этот процесс может быть повторен несколько раз, до тех пор пока не идентифицируется активная гранула.
Прикрепление меток
В этом процессе две молекулы связываются с одной гранулой. Одна из них – новая структура, которая тестируется, тогда как другая является молекулярной меткой (обычно пептид или олигонуклеотид). Эта метка ведет себя, как код для каждой стадии синтеза. В этом процессе гранула должна иметь множественный линкер, способный связываться как с синтезируемой структурой, так и с молекулярной меткой. Реагент присоединяется к одной части линкера, а закодированная аминокислота (или нуклеотид) – к другой части. После каждой последующей стадии комбинаторного синтеза, аминокислота или нуклеотид добавляется к метке, чтобы показать, какой регент применялся.
В качестве примера множественного линкера может привести так называемый safety catch linker (SCAL), который включает в себя остатки лизина и триптофана.
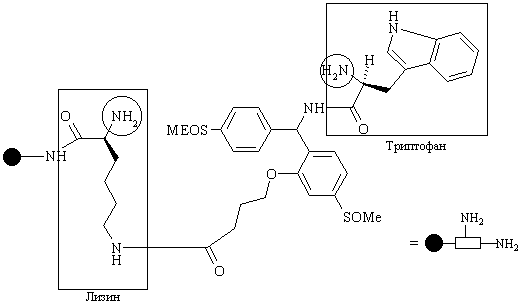
Синтезируемое соединение связывается с триптофановым остатком, а после каждой стадии синтеза меченая аминокислота соединяется с лизиновым остатком и, таким образом, в конце синтеза присутствует трипептид, в котором каждая аминокислота определяет идентичность изменяемых R, R’, R” в непетидной структуре.
Непептидная структура может быть разрушена восстановлением двух сульфоксидных групп в SCAL, который затем взаимодействует с кислотой. В этих условиях порядок трипептидых остатков, соединенных с гранулой, остается неизмененным и может быть установлен для идентификации строения соединения, которое было отделено от гранулы.
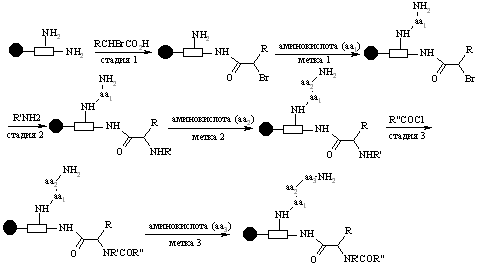
Подобная стратегия может быть использована с олигонуклеотидами вместо меченой молекулы.
Кодировочные таблицы
Разработан метод, который позволяет осуществить разделение индивидуальных твердофазных продуктов, включающий в себя расшифровку кода для определения синтетической истории. Гранулы смолы закрепляются между двумя плетеными листами из инертного полипропилена. Эти листы могут быть промаркированы в виде квадратов и каждый квадрат получает кодировку в виде трех букв. Для примера на рисунке приведены три таких листа размером 6 на 6 см, которые имеют девять маркированных квадратов, каждый из которых получил трехбуквенный код. Эти три листа разделяются и каждый из них обрабатывается аминокислотой. В результате все гранулы верхнего листа связываются с лейцином, все гранулы второго листа связываются с серином, а все гранулы нижнего листа связаны с глицином. Все листы затем промываются, сушатся и обрабатываются пиперидином для удаления Fmoc защитной группы.
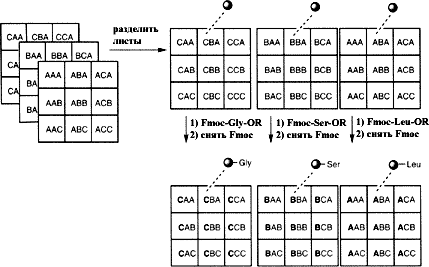
На второй стадии листы разрезаются на три колонки. Каждая колонка обрабатывается затем другой аминокислотой активированной Fmoc и, таким образом, генерируется уникальный дипептид на каждой колонке материала.
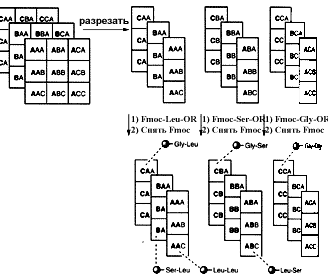
Эти колонки обрабатываются для отделения Fmoc защитной группы. После этого колонки разрезаются на индивидуальные квадраты. Таким образом получаются три набора квадратов. Каждый из этих квадратов обрабатывается третьей аминокислотой и, таким образом, синтезируется 27 возможных трипептидов. Каждый из квадратов содержит уникальный трипептид, который идентифицируется с помощью трехбуквенного кода, имеющегося на каждом квадрате.
Планирование и дизайн комбинаторного синтеза
Паукообразные молекулы
Для нахождения новых лидеров методом комбинаторного синтеза необходимо генерировать большое число различных структур.
В общем, лучше всего синтезировать “паукообразные” молекулы, называемые так, потому что они содержат центральный фрагмент (называемый центроидом или подпоркой (scaffold)), который имеет различные щупальца (заместители).
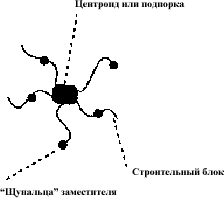
Эти щупальца содержат различные функциональные группы, которые могут быть использованы для нащупывания положений мишени, с которыми эта паукообразная молекула может вступить во взаимодействие. Шанс успеха повышается, если щупальца равномерно распределены вокруг центроида и ощупывают все трехмерное пространство вокруг молекулы.

Еще более успешным может быть получение молекул, которые имеют различные функциональные группы, находящиеся на различном расстоянии от центроида.
Дизайн молекул лекарств
Паукообразные молекулы повышают шанс нахождения лидера, который будет взаимодействовать с мишенью рецептора или энзима, но при этом необходимо хорошо понимать, что соединение, обладающее хорошим взаимодействием с мишенью, не обязательно должно быть хорошим лекарственным препаратом. Для этого необходимо исследование фармокинетики. Существенным ограничением для молекул, получаемых комбинаторным синтезом, является то, что они должны быть активными при оральном приеме. В общем, шансы появления оральной активности повышаются при учете следующих структурных особенностей:
- Молекула должна иметь молекулярную массу меньше, чем 500
- Рассчитанный log P должен быть меньше +5
- Она должна иметь не больше, чем пять групп доноров водородных связей
- Иметь не больше чем 10 групп, акцепторов водородной связи
Группы должны легко подвергаться метаболическим превращениям (например, сложноэфирные). Нужно избегать присутствия в центроиде или заместителях группировок, которые могут привести к токсическим соединениям.
Варьирование заместителей
Набор заместителей, используемых в комбинаторном синтезе, определяется их доступностью и требованиям создаваемого молекулярного множества. Набор заместителей ограничивают такие требования как строение, размер, форма, липофильность, дипольный момент, электростатический заряд, наличие функциональных групп.
Определение активности
Скрининг на грануле
Иногда структуры удается тестировать прямо на твердой фазе. Такой скрининг на грануле предполагает взаимодействие с мишенями, которые мечены ферментами, флюоресцирующей меткой, радионуклидами или хромофорами. Положительная реакция регистрируется по флюоресценции или изменению окраски. Такой метод является очень быстрым и дает возможность анализировать 108 гранул. Активные гранулы могут быть отсортированы микроманипуляцией с последующим установлением строения активного соединения. Однако, может наблюдаться так называемая фальшивая отрицательная активность из-за пространственных трудностей взаимодействия твердой фазы с мишенью. Поэтому все же надежнее удалять синтезированную молекулу с гранулы. Однако иногда бывает, что соединение становится нерастворимым в пробе и дает отрицательный результат, тогда как оно давало положительный эффект находясь на твердой фазе.
Примеры комбинаторного синтеза
Комбинаторная химия удовлетворяет требованиям быстрого получения новых лидеров в различных областях. Большинство ранних работ по комбинаторной химии были в области получения пептидов с использованием твердофазных процедур. Это привело к получению новых ингибиторов протеаз HIV, антимикробных препаратов, лигандов опиатных рецепторов и ингибиторов протеаз аспартамовой кислоты. Однако, пептиды не идеальные кандидаты для лекарственных средств, они обычно обладают слабой оральной активностью, поскольку неустойчивы для пищеварительных ферментов.
Первым опытом получения иных соединений явилась попытка использования таких процедур для получения пептидов на основе неприродных аминокислот. Кроме того, пептиды могут быть модифицированы путем различных превращений, например реакциями N‑метилирования. Пептиды могут быть получены также связыванием N‑замещенных глицинов, давая продукты, которые известны как пептоиды, в которых боковые цепи связаны с азотом, а не с α‑углеродом. Некоторые из них могу служить лигандами для различных важных рецепторов и демонстрируют повышенную метаболическую стабильность.
Ациклические библиотеки
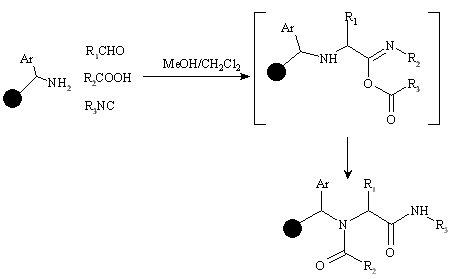
Кроме библиотек пептидов и пептидоподобных структур в ряду ациклических соединений в качестве примера можно привести библиотеку на основе четырехкомпонентной реакции Уги. Синтезирована методом ОГОС на смоле Ринка 96-членная библиотека N-ациламиноалкиламидов из четырех компонентов: 32 карбоновых кислот, одного амина, восьми альдегидов и одного изоцианида:
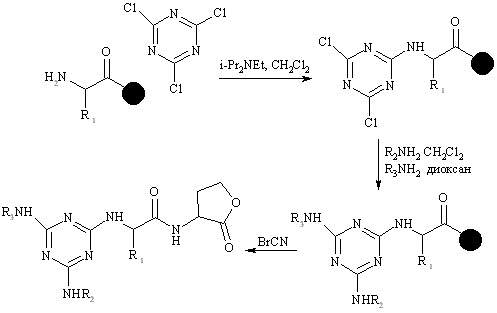
Описана библиотека 12000 триазинов. Избыток цианурхлорида реагирует с иммобилизованнм амином с образованием замещенного хлортриазина. Затем второй атом хлора замещается на аминогруппу, а третий, менее реакционно способный, атом хлора замещается при повышенной температуре.
Синтез пирролидинов
Нанесенные на полистирольные гранулы аминокислоты конденсируются с ароматическими и гетероароматическими альдегидами в триэтилформиате до соответсвующих полимерносвязанных иминов, которые путем циклоприсоединения с олефиновыми и ацетиленовыми диполярофилами дают производные пирролидина и пирролина:
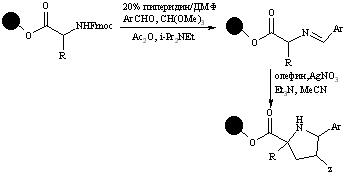
Таким образом получена библиотека из 500 соединений.
Синтез индолов
Приведен синтез аналогов индолов через интрамолекулярную реакцию Хека полимерносвязаных арилгалогенидов.
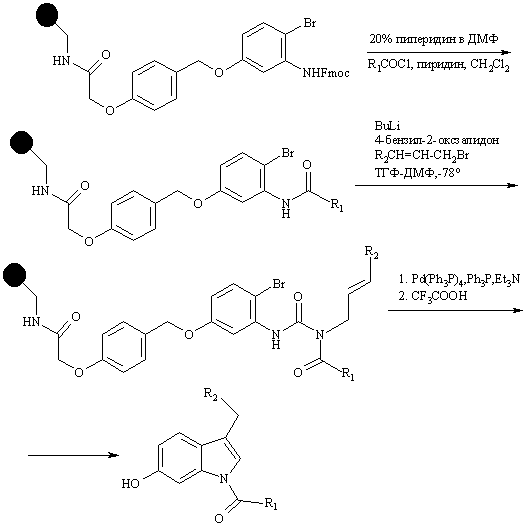
Используются каталитические количества Pd(Ph3P)4 c трифенилфосфином в диметилацетамиде при 85о с последующим снятием индолов с подложки трифторуксусной кислотой.
Передача нервного импульса
Основной единицей нервной системы является нейрон. Нейрон – нервная клетка, функции которой состоит в распространении и интерпретации информации.
Элементарным проявлением активности служит возбуждение, происходящее в результате изменения полярности мембраны нервной клетки. Фактически нервная деятельность является результатом процессов, происходящих в синапсах – в местах контакта двух нейронов, где происходит передача возбуждения от одной клетки к другой. Передача осуществляется с помощью химических соединений – нейромедиаторов. В момент возбуждения значительное количество молекул высвобождается в синаптическую щель (пространство, разделяющее мембраны контактирующих клеток) диффундирует через нее и связываются с рецепторами на поверхности клеток. Последнее и означает восприятие сигнала.
Специфичность взаимодействия нейромедиаторов в рецепторах определяется строением как рецепторов, так лигандов. Основой действия большинства химических веществ на центральную нервную систему является их способность изменять процесс синаптической передачи возбуждения. Чаще всего эти вещества выступают в роли агонистов (активаторы), они повышают функциональную активность рецепторов, или антагонистов (блокаторы). В синапсах нервно-мышечных соединений основным медиатором является хлорацетилхолин. Если нервные узлы расположены вблизи спинного мозга медиатором является норадреналин.
В большинстве возбужденных синапсах в мозге млекопитающих выделяемым нейромедиатором является L‑глутаминовая кислота (1‑аминопропан‑1,3‑дикарбоновая кислота).
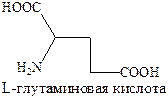
Это один из медиаторов относящийся к классу возбуждающих аминокислот, а γ‑аминомасляная кислота (ГАМК), как и глицин, являются тормозящим медиатором центральной нервной системы. Важнейшие физиологические функции γ‑аминомасляной кислоты – регуляция возбудимости мозга и участие в формировании поведенческих реакций, например, подавление агрессивного состояния.
γ‑аминомасляная кислота образуется в организме путем декарбоксилирования L‑глутаминовой кислоты под действием фермента глутаматдекарбоксилазы.
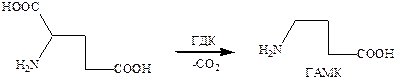
Основной путь метаболического превращения γ‑аминомасляной кислоты в нервной ткани – это трансаминирование с участием α‑кетоглутаровой кислоты. Катализатором в этом случае служит фермент ГАМК-Т (ГАМК-трансамилаза). Трансаминирование приводит к глутаминовой кислоте, метаболическому предшественнику γ‑аминомасляной кислоты и янтарному полуальдегиду, превращающегося затем в ГОМК (γ‑оксимасляная кислота), которая является антигипоксическим средством.
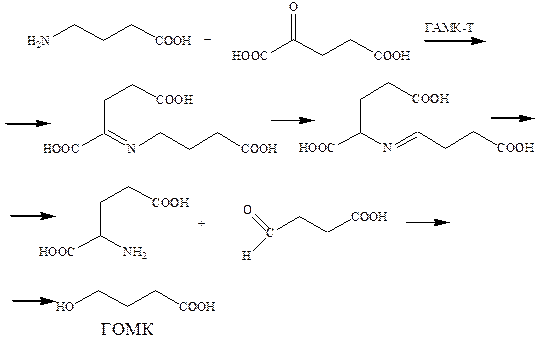
Именно этот процесс инактивации γ‑аминомасляной кислоты стал целевым для исследований, направленных на накопление медиаторов в тканях мозга, для усиления его нейротормозной активности.
Считается, что 70% центральных синапсов предназначенных для стимуляции центральной нервной системы используют в качестве медиатора L‑глутаминовую кислоту, а вот избыточное накопление его приводит к необратимым повреждениям нейронов и тяжелым патологиям типа болезни Альцгеймера, инсульта и т.д.
Глутаматные рецепторы делятся на два основных типа:
1. ионотропные (i Gly Rs)
2. метаботропные (m Gly Rs)
Ионотропные глутаматные рецепторы образуют ионные каналы и непосредственно передают электрический сигнал от нервных клеток за счет возникновения ионного тока.
Метаботропные глутаматные рецепторы переносят электрический сигнал не непосредственно, а через систему вторичных мессенджеров – молекулы или ионы, которые в итоге вызывают изменения конфигурации белков, участвующих в специфических клеточных процессах.
Ионотропные глутаматные рецепторы – семейство глутаматных рецепторов, связанных с ионными каналами. Включает в себя два подтипа, различающихся по фармакологическим и структурным свойствам. Название этих подтипов образованы от названий наиболее селективных лигандов-агонистов к каждому из соответствующих рецепторов. Таковыми являются N‑метил‑D‑аспарагиновая кислота (NMDA), 2‑амино‑3‑гидрокси‑5‑метилизоксазол‑4‑ил‑пропановая кислота (AMPA), каиновая кислота
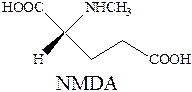
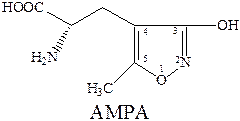
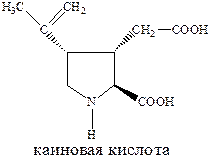
Таким образом различают два подтипа ионотропных глутаматных рецепторов: NMDA и NMPA (каинатный подтип).
NMDA наиболее изученный из всех глутаматных рецепторов. Исследования действия соединений различных классов показало наличие в нем несколько сайтов регуляций – это область специального связывания с лигандами. Рецептор NMDA имеет два аминокислотных сайта: один для специфического связывания глутаминовой кислоты, другой для специфического связывания глицина, являющиеся коагонистами глутамата. Иными словами, для открытия ионного канала необходима активация обоих (глутаминового и глицинового) связывающих центров. Канал сопряженный с рецепторами NMDA проницаем для катионов Na+, K+, Ca2+ и именно с увеличением внутриклеточной концентрации ионов кальция связывают гибель нервных клеток при заболеваниях, сопровождающихся гипервозбуждением рецептора NMDA.
В канале рецептора NMDA существует сайт специфического связывания двухвалентных ионов Mg2+ и Zn2+, которые оказывают ингибирующее действие на процессы синаптического возбуждения рецепторов NMDA. На рецепторе NMDA присутствуют и другие аллостерические модуляторные сайты, т.е. такие, взаимодействие с которыми не оказывает прямого действия на основную медиаторную передачу, но способны влиять на функционирование рецептора. Таковыми являются:
1) Фенциклидиновый сайт. Он расположен в ионном канале, а действие фенциклидина заключается в селективном блокировании открытого ионного канала.
2) Полиаминовый сайт, расположенный на внутренней стороне постсинаптической мембраны нейрона и способный связывать некоторые эндогенные полиамины, например, спермидин, спермин.
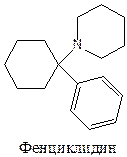
Рассмотрим химию соединений активных по отношению к рецепторам NMDA.
Агонисты
Молекула AMPA представляет собой аналог глутаминовой кислоты, в которой роль терминальной карбоксильной группы играет кислая гидроксиизоксазольная группа. Активность к соответствующему рецептору проявляет только S‑изомер AMPA, в то время как R‑изомер практически не активен. Замена атомов водорода метильной группы на галоген (-СF3, -CH2Cl) приводит к агонистам с активностью близкой к активности AMPA. Однако введение объемной третбутильной группировки приводит к потере агонистической активности по отношению к каинатному рецептору. Замена гидроксильной группы в изоксазоле на карбоксильную группировку приводит к пятикратному повышению активности как антагониста.
К сильным агонистам AMPA относятся и природный β‑оксозиламино‑L‑аланин.
Структуры известных в настоящее время агонистов каиновых рецепторов довольно близки к самой каиновой кислоте. Интересным примером конструирования агониста AMPA каинатного рецептора является объединение в одной молекуле фрагмента AMPA и каиновой кислоты.
Производные пролина обладают слабой агонистической активностью по отношению к рецептору AMPA, но также способно к связыванию с рецептором AMDA.
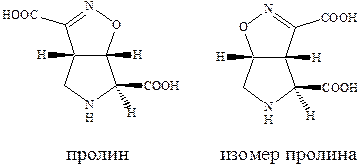
Изомер пролина является высоко аффинным и активным агонистом AMPA каинатных рецепторов, хотя, как можно было ожидать, и не слишком селективным по отношению к каждому из подтипов. Очень сильным и очень селективным лигандом каинатного рецептора является такое простое соединение как 4‑метилглутаминовая кислота, которая в 3000 раз более селективна к каинатному, чем к рецептору AMPA и в 200 раз более селективна к каинатному рецептору, чем рецептору NMDA.
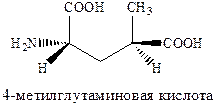
Структурные требования к выбору селективных агонистов сайта связывания глутаминовой кислоты AMPA каинатных рецепторов, отличающих их от таковых для рецепторов NMDA, не вполне ясны за исключением предпочтительности S‑конфигурации хирального аминокислотного центра.
Противомикробные препараты
Наиболее часто причиной заболевания человека и животных является проникновение в организм болезнетворных возбудителей, а именно: вирусов, микробов, грибков, простейших, гельминтов (паразиты, например, бычий цепень, аскарида), а также перерождение нормальной клетки в раковую.
Для лечения таких заболеваний используют лекарственные препараты, действующие на болезнетворного возбудителя или раковую клетку. В зависимости от того, на какой возбудитель действует лекарственный препарат, различают противомикробные, противопаразитарные, противовирусные, противогрибковые и препараты для лечения злокачественных новообразований. Отметим, что борьба с болезнетворными бактериями это один из наиболее успешных разделов медицинской химии. Впервые идентификация болезнетворных бактерий осуществлена Ван Левенгуком после открытия микроскопа в 1870 году. В середине XIX века были выявлены бактерии ответственные за холеру, тиф, туберкулез.
Использование бактерий в роли бактериологического оружия представляет собой страшную силу, способную не менее эффективно, чем химическое, уничтожать огромную массу людей.
В соответствии с предложенной Грамом систематикой, бактерии делятся на два типа:
- грамположительные, окрашиваемые в синий цвет смесью красителей кристаллического фиолетового с иодом (реактив Грама).
- грамотрицательные, не окрашиваемые реактивом Грама.
Грамотрицательные бактерии более устойчивы к бактерицидным препаратам, чем грамотрицательные, и для борьбы с ними необходим более сильнодействующий препарат.
Противомикробные препараты – это прежде всего сульфаниламиды. Их история начинается с 1935 года, когда впервые были проведены их испытания in vivo (внутри организма).
Сульфаниламиды обладают химиотерапевтической активностью при инфекциях, вызванных грамположительными и грамотрицательными бактериями.
Основной представитель – белый стрептоцид (парааминобензолсульфонамид).
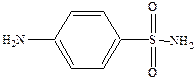
Его водорастворимая форма п‑сульфонамидфениламинометансульфонат натрия.
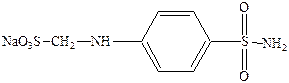
Белый стрептоцид имеет место быть в организме в результате метаболизма красного стрептоцида.
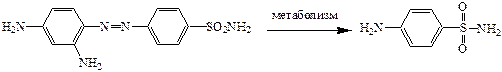
Но из-за токсичности красный стрептоцид снят с производства.
Механизм действия сульфаниламидов выяснен на уровне ферментов (досконально). Антибактериальная активность стрептоцида связана с ингибированием процесса синтеза дигидрофолиевой кислоты – важного фактора роста микроорганизмов. В этих процессах патогенные для человека бактерии используют в качестве строительного блока природный метаболит – п-аминобензойную кислоту.
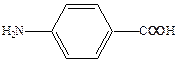
Пользуясь структурным сходством с п-аминобензойной кислотой, сульфаниламиды конкурентно включаются в процесс превращения пирофосфата в ложную дигидрофолиевую кислоту. Последняя не способна выполнить свою жизненную функцию и губительна для микроорганизмов. Дигидрофолиевая кислота под действием восстанавливающего фермента превращается в тетрагидрофолиевую кислоту, которая служит переносчиком углеродных фрагментов и играет важную роль в живых организмах как млекопитающих так и бактерий.
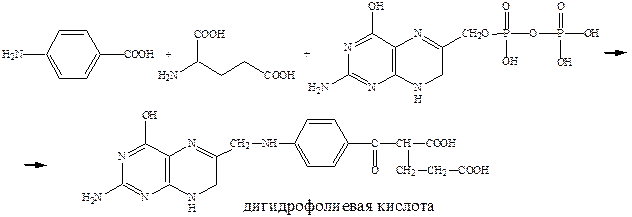
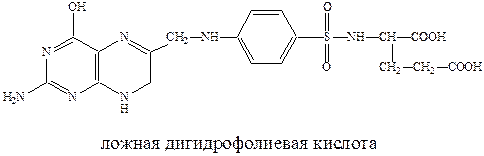
Для бактерий процесс синтеза дигидрофолиевой кислоты на основе п-аминобензойной кислоты является очень важным. Бактерии, в отличие от млекопитающих, могут получать этот продукт только в результате синтеза. А, например, люди к синтезу не способны и получают ее с пищей. Отсюда ингибирование этого синтеза приводит к гибели патогенных бактерий, не затрагивая жизненноважных функций организма млекопитающих.
Сульфаниламиды близки по структуре, электронным свойствам и размерам молекулы к п-аминобензойной кислоте. Вмешиваясь в синтез на стадии конденсации п-аминобензойной кислоты с глутаминовой кислотой и пиридиновым основанием сульфаниламиды нарушают синтез тимино- и пуриновых оснований, что ведет к быстрой гибели бактерий, лишенных возможности синтеза собственных ДНК.
Структурные аналоги метаболитов, подавляющие их биологические функции, называются антагонистами или антиметаболитами. Таким образом, антибактериальное действие сульфаниламидов обусловлены тем, что они являются антиметаболитами. Понятно, что многое решают дозы, в которых они используются, так как блокада ферментов является конкурентной и надо применять такие количества лекарственного препарата, которые исключат возможность использования микроорганизмами п-аминобензойной кислоты.
Недостаточные дозы или прерванный курс лечения приведут к выработке штаммов возбудителей резистентных (устойчивых) к сульфаниламидам.
Выявление физиологической активности синтезируемых структурных аналогов сульфаниламидов позволило дать заключение:
- Основная для проявления активности п-аминогруппа, она должна быть не защищенной (за исключением ацильной защиты
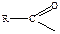 ).
). - Необходимо присутствие сульфамидной группы в ароматическом ядре.
- В ароматическом кольце должны быть замещены только пара‑положения.
- Азот-сульфамидные группы должны быть вторичными, и только радикал при нем может варьироваться.
Эти особенности выявлены в результате молекулярного дизайна.
Ряд сульфаниламидных препаратов получен за счет модификации сульфамидной группы, при этом имеет место наиболее целенаправленное действие. Так сульфацил натрий (альбуцид) широко применяется для лечения глазных болезней.
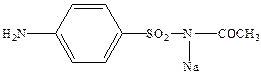
Ряд сульфамидных препаратов получен путем введения сульфамидную группу остатков гетероциклических оснований. К таким препаратам можно отнести сульфодиметоксин – лекарство длительного действия, и сульфален – сверхдлительного действия.
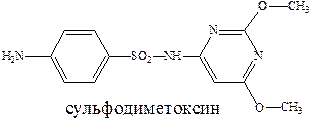
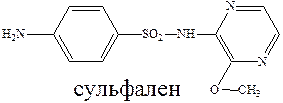
Введение дополнительных гетероциклических заместителей позволяет пролонгировать действие лекарственного препарата, улучшить проникновение их в жидкости и ткани организма и тем самым увеличить эффективность их действия.
Антибиотики
Антибиотики – продукты нормального обмена веществ любых живых организмов, способные убивать бактерии, грибы, вирусы. Их открытие связано с именем английского ученого Флеминга, впервые обнаружившего противомикробную активность зеленой плесени в 1928 году. А выделение действующего начала зеленой плесени (натриевой соли пенициллина) осуществил английский ученый Флори в 1940 году. Введение в медицинскую практику антибиотиков позволило эффективно лечить пневмонию, менингит, сепсис, ангину, кишечные инфекции, холеру, дизентерию, туберкулез и др.
Антибиотики имеют различное химическое строение.
Первая группа антибиотиков – β‑лактамные (пенициллины), эффективны при инфекциях вызванных грамположительными бактериями (стрептококки, стафилококки, пневмококки). Пенициллины оказывают выраженный эффект в отношении растущих микроорганизмов (бактерицидное действие). Они поражают бактерии в фазе роста, ослабляя их клеточные стенки, а учитывая, что для бактерий характерно необыкновенно высокое внутреннее давление, это приводит к разрыву клеточной стенки и уничтожению бактерии. Активность препаратов пенициллина определяют по антибактериальному действию на определенный штамм золотистого стафилококка. За единицу действия принимают активность 0,5988 г химически чистой кристаллической натриевой соли бензилпенициллина.
Результат скрининга «структура‑свойства» для пенициллина сводится к следующему:
- Обязателен лактамный цикл (внутренний амид).
- Обязательно наличие свободной карбоксильной группы
- Важна бициклическая система.
- Важна ациламинная связь.
- Часто сера необязательна.
- Цис-стереохимия обязательна в бициклической системе.
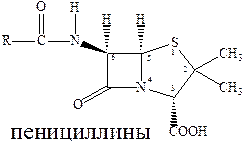
Серьезной проблемой, стоящей на пути использования пенициллина, кроме их малой кислотоустойчивости, является и возникновение резистентности – для пенициллинов это общая проблема, касающаяся и многих других препаратов. Например, микробактерии туберкулеза обладают природной резистентностью к пенициллину, а многие штаммы стафилококка, чувствительные к действию антибиотиков, достаточно легко приобретают резистентность к ним.
Важным механизмом такой резистентности микроорганизмов по отношению к β‑лактамным антибиотикам является образование бактериями ферментов – β‑лактамаз, которые раскрывают β‑лактамные циклы и тем самым лишают эти антибиотики возможности выступать в качестве ацилирующего агента, что и является основой их биологического действия. При этом пенициллин превращается в неактивное производное пенициллиновой кислоты.
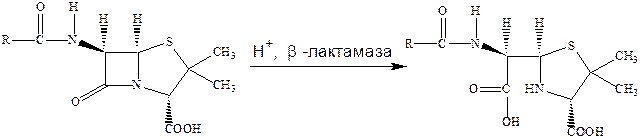
Главный путь борьбы с этим явлением – поиск соединений, способных ингибировать эти ферменты и предотвращать инактивацию антибиотиков. Было установлено, что таким эффективным ингибитором β‑лактамаз является клавулановая кислота.
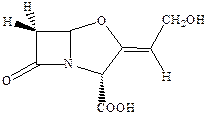
Сама по себе она не обладает антибактериальной активностью и применяется только с β‑лактамными антибиотиками. Интересно, что пенициллины являются синергистами стрептомицина (противотуберкулезный лекарственный препарат, относящийся к аминогликозидам).
Синергизм – одновременное комбинированное воздействие двух (или более) факторов, характеризующихся тем, что такое совместное действие значительно превосходит эффект каждого отдельно взятого компонента.
Скелет молекулы пенициллина наводит на мысль о том, что пенициллин состоит из аминокислот цистеина и валина, это может быть установлено с помощью меченых атомов. Пенициллин ингибирует синтез клеточной стенки, ослабляет мембрану бактериальной клетки и облегчает доступ стрептомицина не только к внеклеточным, но и внутриклеточным микобактериям туберкулеза.
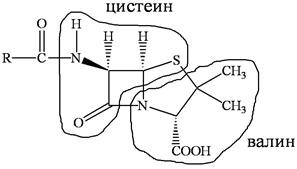
У пенициллинов есть и минусы. Их отличает узкий спектр действия, и, что более существенно, они не подавляют грамотрицательные бактерии.
Учитывая, что пенициллин способен вызывать аллергическую реакцию организма, вполне понятны попытки синтеза аналогов, не обладающих аллергической реакцией. Сюда входит группа цефалоспоринов во многом сходных структурно с пенициллином. Они также содержат β‑лактамное кольцо, но здесь оно аннелировано (аннелирование – процесс пристраивания карбоцикла или гетероцикла к уже существующему в исходной молекуле циклическому фрагменту) не с тиазолидиновым, а с 3,4‑дигидро‑1,3‑тиозиновым циклом.
Цефалоспорины также проявляют бактерицидную активность за счет блокады D‑аланин‑узнающей полимеразы, ответственной за синтез новой клеточной оболочки бактерий. Пенициллины – антагонисты D‑аланил‑D‑аланинов. Главная особенность цефалоспоринов по сравнению с пенициллинами большая устойчивость первых по отношению к β‑лактамазам. Цефалоспорины обладают более широким спектром действия, включая влияние на грамотрицательные микроорганизмы.
Американская фирма Lilly впервые продемонстрировала синтез лекарства, в котором пятичленное тиазолидиновое кольцо пенициллина превращается в шестичленное дигидротиозиновое кольцо цефалоспорина.
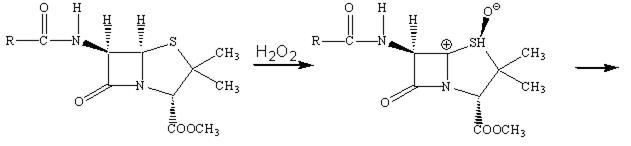
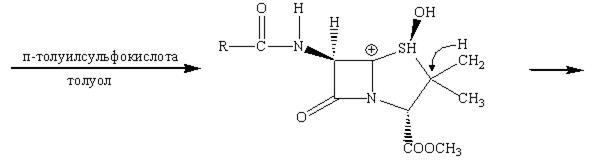
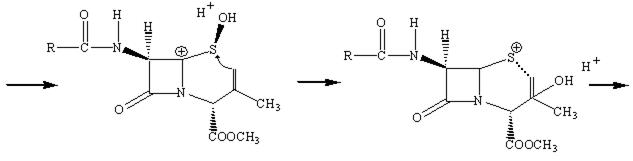
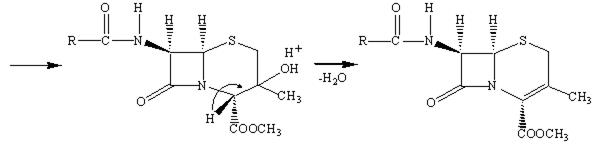
Наличие метильной группы в положении 3 для ряда цефалоспоринов обычно плохо сказывается на их активности, но значительно облегчает всасывание этих препаратов кишечником.
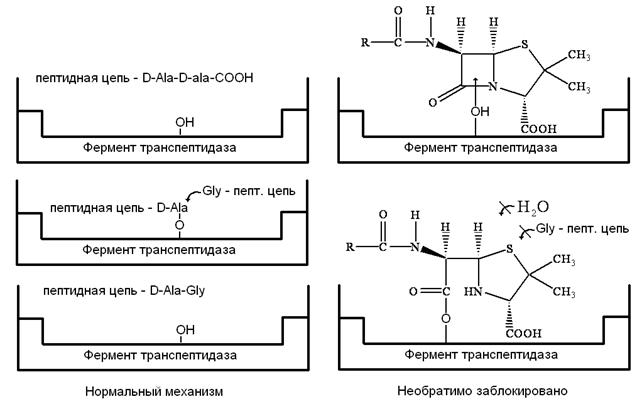
Рассмотрим механизм действия β‑лактамных антибиотиков. Как мы уже отмечали, для бактериальных клеток характерно необыкновенно высокое внутреннее осмотическое давление. От разрыва их удерживает стенка, прочность которой придает пептидогликал. В молекуле пептидогликала концевой всегда является пара D‑аланил‑D‑аланин (D‑Ala‑D‑Ala). Было предположено, что конформация пенициллина подобна таковой, если взять часть сложной аминокислотной цепочки D‑аланил‑D‑аланина при ее поперечном связывании пока это есть реакционный центр для фермента (т.е. мишень). Транспептидаза ошибочно допускает молекулу пенициллина вместо D‑аланил‑D‑аланина в активный сайт и реакция в активном сайте протекает уже с ним. В нормальном механизме амидная связь между двумя аланиновыми звеньями пептидной цепи расщепляется, и последнее звено аланина уходит из пептидной цепи в активном сайте. В пентаглицине последний фрагмент глицина может вступить в сайт и образовать пептидную связь с фрагментом аланина и таким образом удалиться из активного сайта.
Фермент может атаковать β‑лактамное кольцо пенициллина и открыть его также, как он делает это с амидной связью пептида. Поскольку пенициллин имеет циклическое строение, он не расщепляется на две части и не покидает активный сайт. Последующий гидролиз ацильной группы не происходит, т.к. глицин не способен вытащить из сайта объемную молекулу пенициллина.
Противовирусные препараты
Противовирусных препаратов чрезвычайно мало. Это объясняется недостаточными знаниями природы вирусов.
Вирусы – внеклеточная форма жизни, структурно более простая, чем бактерии. Существующий вне клетки вирус (вирион) содержит ядро, состоящее из ДНК или РНК, покрытой защитной оболочкой (капсид), построенной из одного или двух белков. В зависимости от природы нуклеиновой кислоты различают ДНК- и РНК‑вирусы.
Вирусы могут различаться по числу генов, входящих в состав нуклеиновой кислоты. Вирусы несут крайне ограниченное количество генетической информации. Их белковая оболочка содержит большое число белковых субъединиц одного или нескольких видов. Эта белковая оболочка надежно защищает нуклеиновую кислоту вируса от действия нуклеаз. Она также способна внедриться в клеточную стенку бактерий, после чего ДНК обнажается и проникает через клеточную мембрану.
В результате попадания вирусной ДНК или РНК в клетку, останавливается синтез нуклеиновых кислот клетки хозяина всего через несколько минут и начинается синтез вирусных макромолекул. После заражения клетки хозяина, размножение вируса может привести к лизису (разрушение клеточных стенок под действием ферментов). Кроме того, ДНК вируса может включаться в ДНК клетки и вызывать трансформацию клетки, в том числе и в раковую, поэтому при создании антивирусных нужно учитывать все факторы химического строения и развития вируса.
Действие противовирусного препарата должно быть направлено на какой-то этап вирусной инфекции: проникновение вируса в клетку, депротеинизация, синтез специфических белков, и при всем этом препарат должен быть не токсичен для хозяина на всех уровнях его применения. Отсюда основной подход к поиску антивирусных препаратов – это строгая направленность, основанная на определенные мишени – вирусный геном, вирус‑содержащие ферменты, ДНК- и РНК‑полимеразы вирусов.
Противовирусным эффектом обладают производные адамантана (ремантадин) и оксалин.
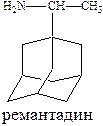
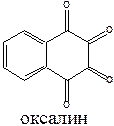
Противовирусным действием обладают также интерфероны (эндогенные низкомолекулярные белки, моль масса которых колеблется в пределах от 15 до 25 тысяч), продуцируемые в клетках человека в ответ на внедрение вируса. Они нарушают процесс репликации вирусов и являются одним из факторов неспецифического иммунитета, делая клетки частично или полностью резистентными по отношению к вирусным инфекциям.
Однако самым важным в случае такой инфекции как герпес является препарат ацикловир.
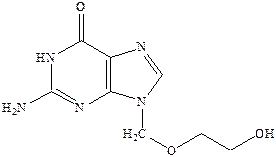
Ацикловир применяется в пораженных вирусом клетках, где он превращается в моно‑, а затем ди‑ и трифосфат, который тормозит синтез и репликацию ДНК вируса.
Зидовудин (азидотимидин) оказывает иммуностимулирующее действие в комплексной терапии СПИДа, поскольку подавляет репликацию ВИЧ, но не убивает.
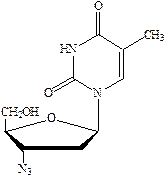
Противоопухолевые препараты
Злокачественные новообразования являются одной из ведущих проблем, ибо ежегодно свыше 5 миллионов человек на Земле умирают от этой патологии. Известно несколько групп соединений, которые проявляют противоопухолевый эффект, но при этом, как правило, затрагивают и нормальные клетки. Это антиметаболиты и цитостатические препараты: ацилирующие агенты, комплексы лигандов, природные соединения алкалоиды, антибиотики, ферменты и гормоны.
Действие антиметаболитов обусловлено нарушением синтеза нуклеиновых кислот. Они разнообразны по структуре. Обычно это производные витаминов, гетероциклических оснований, кислот и т.д. Они влияют на активность фермента, что проявляется в виде терапевтического эффекта.
Большинство простейших организмов не способны синтезировать пурины и должны получать их от организма хозяина и должны иметь ферментативные системы пригодные для их переработки. Отсюда, поиск соответствующих антиметаболитов.
К числу антиметаболитов аналогов пурина относят 6‑меркаптопурин. Механизм цитостатического действия его обусловлен нарушением синтеза ДНК и РНК путем блокады включения в них аденина и гуанина. Будучи структурным аналогом аденина 6‑меркаптопурин является антиметаболитом пурина. Он нарушает нормальный пуриновый обмен, вследствие чего нарушается синтез нуклеиновых кислот. Препарат показан при лечении острого лейкоза (рак крови).
Антиметаболиты, производные пиримидина, нарушают пиримидиновый обмен, влияют на активность фермента, участвующего в синтезе нуклеиновых кислот. Например, 5‑фторурацил снижает активность фермента тимидил-синтетазы.
5‑фторурацил это пример образования высокоактивных соединений при метаболизме менее эффективного противоопухолевого препарата, который называется фторафур.
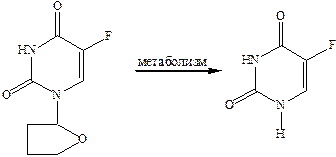
Его применяют при рецидивном и иноперабельном раке желудка, толстой и прямой кишки, поджелудочной железы. 5‑фторурацил обладает сильным токсическим действием.
Наиболее многочисленна группа цитостатических препаратов, включающая ацилирующие агенты, например,
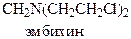
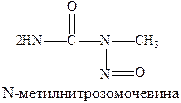
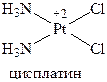
Цисплатин представляет собой плоский комплекс с центральным атомом платины, окруженным двумя лабильными атомами хлора и двумя относительно инертными аммониевыми фрагментами в цис‑конфигурации.
Ацилирующие агенты способны реагировать с нуклеофильными центрами белковых молекул, нарушая главным образом синтез ДНК и в меньшей степени синтез РНК. В результате нарушается жизнедеятельность клетки.
Хлорэтиламины обладают высокой токсичностью и в дозах, близких к лечебным, могут вызывать побочные явления проявляющиеся в сильном угнетении костно-мозгового кроветворения и нарушениях функции желудочно-кишечного тракта. Эмбихин применяют при лимфогрануломатозе, лимфолейкозе, а N-метилнитрозомочевину при раке легких.
Цисплатин по механизму действия сходен с алкилирующими агентами. Главной мишенью для проявления цитотоксичности цисплатина является ДНК. Он взаимодействует именно с пиримидиновыми и пуриновыми основаниями ДНК, не реагируя с фосфатами или сахарными остатками. Сшивая нити ДНК, цисплатин обеспечивает длительное подавление биосинтеза и гибель патологических клеток. Применяют его при злокачественных опухолях мочевого пузыря.
В борьбе с опухолевыми патологиями могут полезны ферменты. Речь прежде всего идет о L‑аспарагиназе – фермент, основная функция которого дезаминирование аспарагина до аспарагиновой кислоты. Вероятный механизм его действия: способность нарушать метаболизм аминокислоты аспарагина, которая необходима некоторым опухолевым клеткам. Иногда L‑аспарагиназа оказывается более эффективной, чем другие противоопухолевые препараты.
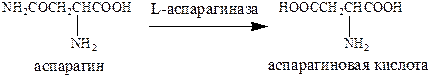
Недавно появились новые данные о возникновение и росте опухолей при воздействии свободных радикалов, в частности оксида азота (II) – N=O. Целый ряд патологических состояний в том числе сердечно-сосудистые заболевания, инфекции, повреждения мозга вызваны избыточным высвобождение N=O. Нежелательными токсикологическими эффектами перепродукции N=O являются коллапс (падение ритмов сердца до нуля), клеточные повреждения, септический шок (падение давления), поэтому в ряде случаев возникает необходимость ингибирования N=O.
Оксид азота при перепродукции его в организме может выступать в качестве медиаторов воспаления и факторов вызывающих модификацию белков и повреждение нуклеиновых кислот. Эти эффекты могут привести к инициализации канцерогенеза. Возможно, именно свободные радикалы и N=O ответственны за повреждение белков, мембран, ДНК и РНК.
Анальгетики
Под анальгетиками подразумеваются средства, доминирующим эффектом которых является ослабление или устранение чувства боли. Их принято разделять на две группы:
а) наркотические анальгетики: морфин и близкие к нему алкалоиды (опиаты) и синтетические соединения, обладающие подобными свойствами (опиоиды).
б) ненаркотические анальгетики, включая нестероидные противовоспалительные препараты.
В обычной медицинской практике широко распространены анальгетики относящиеся к производным пиразола и п‑аминофенола. Производные фенилпиразалола – аминопирин и анальгин. Производные п‑аминофенола – фенацетин и парацетамол. Производные салициловой кислоты – салицилат натрия, аспирин, салициламид.
Обычно эти препараты оказывают не только обезболивающий эффект, но и жаропонижающий, т.е. являются антипиретиками.
В последнее время установлено, что применение аминопирина и фенацетина (если это длительно) вызывает нежелательные побочные эффекты, вплоть до канцерогенного влияния аминопирина и нефротоксического действия фенацетина, что привело к ограничению использования этих препаратов.
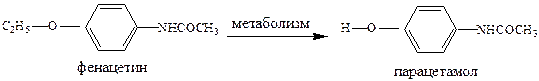
Парацетамол является основным метаболитом фенацетина и дезаминирование в организме протекает очень быстро. По-видимому, обезболивающий эффект фенацетина вызван именно этим обстоятельством, т.е. фенацетин - пролекарство, а парацетамол – истинное лекарство. Парацетамол не обладает противовоспалительной активностью и применяется как болеутоляющее средство. В его анальгетическом эффекте имеется центральный компонент – снижается уровень простагландинов в структурах головного мозга, уменьшается поступление болевых импульсов по восходящим путям спинного мозга.
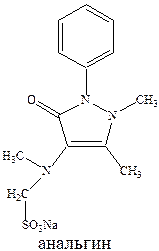
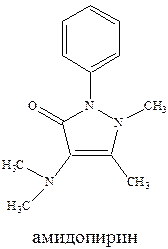
Анальгин обычно применяется для снижения температуры тела. Его эффект развивается быстро в отличие от амидопирина, не обладает судорожным действием, может назначаться перорально, внутривенно, внутримышечно. Однако (и это свойственно для всех производных пирозалола) он резко угнетает кроветворение. В этой связи сейчас все чаще применяют жаропонижающее средство парацетамол, причем чаще применяется его комбинация с ацетилсалициловой кислотой. При этом усиливается и жаропонижающий и болеутоляющий эффекты, и что очень важно, парацетамол защищает слизистую желудка от раздражающего действия ацетилсалициловой кислоты.
Салицилаты – это первые препараты, для которых было обнаружено противовоспалительная активность, но механизм действия салициловой кислоты все еще изучается.
Салициловая кислота оказывает противовоспалительный, жаропонижающий и болеутоляющий эффекты и применяется еще как антиревматическое средство.
Противовоспалительная активность ацетилсалициловой кислоты вызвана влиянием на процессы, протекающие в очаге воспаления, а именно снижением проницаемости капилляров и снижении активности фермента, расщепляющего гликозидные связи, и ингибированием энергетического обеспечения путем торможения образования аденозинтрифосфата. Важную роль в этом отношении имеет ингибирование синтеза простагландинов свойственное для нестероидных противовоспалительных препаратов.
Анальгезирующий эффект связан с действием на центры болевой чувствительности, а также способностью снижать вызывающее боль действие брадекинина (чушь какая-то, абракадабрина). С влиянием на биосинтез, метаболизм и высвобождение простагландинов связаны различные положительные и негативные эффекты ацетилсалициловой кислоты. Именно с этим связано антиагрегационная активность этого препарата – способность ингибировать агрегацию тромбоцитов, что находит применение для предотвращения различных тромбозов: послеоперационных тромбофлебитов, нарушения мозгового кровообращения, образование тромбов при ишемической болезни сердца. С другой стороны, торможение синтеза простагландинов приводит к ряду осложнений, важнейшими из которых являются повышение кислотности желудочного сока, нарушение кровоснабжения слизистой, возникновение желудочных кровотечений. Поскольку такой риск существует не только для ацетилсалициловой кислоты, но и для других нестероидных противовоспалительных средств целесообразно посмотреть данные, связанные с биосинтезом и ролью простагландинов в живом организме.
Простагландины – это вторичные месенджеры, преобразующие информацию гормонов или нейромедиаторов в соответствии физиологическому ответу. Обычно они действуют сильно, но кратковременно, т.к. разрушение из специфическими дегидрогеназами протекает быстро.
Из продуктов питания в организм поступает γ‑линолениковая кислота, которая затем превращается в арахидоновую, накапливающуюся в составе фосфолипидов клеточной мембраны и высвобождающуюся оттуда под действием фермента фосфолипазы.
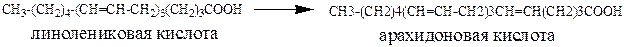
Простогландины играют важнейшую роль в течении физиологических и паталогических процессов в тканях, поэтому при торможении их синтеза в стенке желудка, где простагландины регулируют ее защитные свойства, нормализуя кровоснабжение и снижая секрецию соляной кислоты, приводит к ульцерогенному эффекту (изъязвлению).
Второй путь метаболизма арахидоновой кислоты связан с ее трансформацией под действием ферментов, в результате образуются лекотриены, вызывающие воспаление и ответственные за симптомы астмы.
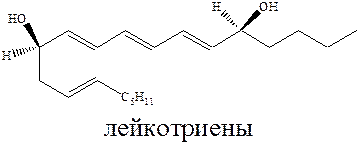
Необратимое ингибирование нестероидными противовоспалительными препаратами простагландинсинтетазы приводит к побочным эффектам, связанными с желудочными заболеваниями. С другой стороны уменьшение синтеза простогландинов приводит к нормализации функции тканей пораженных ревматизмом. Опасным осложнением применения нестероидных противовоспалительных препаратов является бронхоспазм.
Блокада синтеза простагландинов может вызвать сдвиг метаболизма арахидоновой кислоты в сторону лейкотриенов, а это отек тканей и спазмы гладкой мускулатуры.
К числу наиболее эффективных противовоспалительных средств относятся ибупрофен и ортофен.
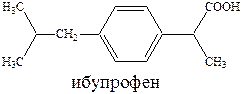
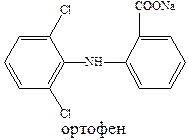
Ибупрофен малотоксичен и близок к салицилатам по противовоспалительной и анальгезирующей активности. Стимулирует образование эндогенного интерферона, обладает значительным жаропонижающим эффектом, лечит ревматоидный артрит и снижает температуру тела.
Ортофен хорошо проникает в полость суставов при ревматоидном артрите, артрозах. Используется для купирования болей и при заболеваниях слизистой оболочки полости рта и парадонтите.
Наркотические анальгетики
Основным источником наркотических анальгетиков являются алкалоиды опия, выделяемые из опийного мака. Синтез морфина впервые был осуществлен в 1950 году, но не нашел промышленного применения в виду своей сложности. Производные морфина кодеин и этилморфин синтезируют из морфина. Более простые заменители морфина промедол и лидол получают путем химических превращений.
Эта группа соединений характеризуется сильной анальгезирующей активностью, позволяющей использовать их в экстраординарных случаях (хирургическом вмешательстве, ранениях, злокачественных новообразованиях, сопровождающихся сильным болевым синдромом).
Другой характеристикой соединений этого типа является влияние на центральную нервную систему, выражающуюся в эйфории и, в конечном счете, появлением физической и психологической зависимости (наркомания), что ограничивает их длительное применение, несмотря на имеющуюся их эффективность.
Развитие синдрома физической зависимости приводит к тяжелым последствиям – синдрому абстиненции («ломка»), при прекращении введения препарата.
Еще одна особенность – наличие специфических антагонистов, способных снять как анальгезирующее действие, так и токсические явления, связанные с применением этих препаратов.
В малых концентрациях, не достаточных для того, чтобы вызвать сон, морфин вызывает эйфорию и освобождает от боли – анальгезию.
Морфин обладает выраженной стереоселективностью.
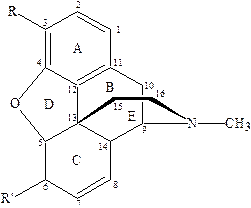 или
или 
В молекуле этого алкалоида 5 асимметрических атомов углерода (С5, С6, С9, С13, С14). Такое количество асимметрических атомов теоретически допускает возможность существования 32 оптических изомеров морфина, но ограничения, которые налагаются мостиковой этиламинной цепочкой, создающей кольцевую систему С9-С13, приводит к тому, что морфин существует лишь в виде 16 оптических изомеров. Центры С5, С6, С9 являются левовращающими, а С13, С14 – правовращающими.
Одной из причин отличающейся физиологической активности стереоизомерных лекарственных препаратов является различия в их проникновении в организм. Они могут быть связаны как с особенностями строения и свойствами биологических мембран, которые сами построены из оптически активного асимметрического материала, так и с наличием в мембранах специальных систем, осуществляющих перенос метаболитов через мембраны.
Морфин и близкие алкалоиды – яркий пример влияния пространственной конформации на физиологическую активность соединения. Морфин, содержащийся в естественном растительном сырье, является одним из левовращающих изомеров. Введение этого препарата вызывает сильную анальгезию, а синтезированный правовращающих изомер морфина полностью лишен каких бы то ни было анальгезирующих свойств.
Молекулу морфина много раз модифицировали. При этом удавалось получать соединения со значительно большей анальгезирующей активностью. К сожалению повышение активности всегда сопровождалось повышением токсичности, способностью вызывать пристрастие, к сокращению продолжительности эффекта и другим нежелательным явлениям.
| R | R’ | Активность, в относительных единицах | Название препарата |
| НО | НО | 100 | морфин |
| CH3O | НО | 10 | кодеин |
| C2H5O | НО | 10 | дионин |
| НО | CH3O | 160 | гетерокодеин |
| НО | OCOCH3 | 400 | α‑ацетилморфин |
| OCOCH3 | OCOCH3 | 180 | диаморфин, героин |
Токсичность препарата оценивается дозой LD50, при введении которой наступает гибель 50% экспериментальных животных.
Анальгин обладает значительно меньшими анальгезирующими свойствами, чем морфин, но часто достаточными, чтобы помочь при болях. Кроме того, кодеин оказывает угнетающее действие на кашлевой центр, тем самым снижается частота кашля. Кодеин в меньшей степени чем морфин, но вызывает зависимость. Отсюда, для его применения те же ограничения, что и для других наркотических анальгетиков.
При передозировке морфина имеет место угнетение дыхания и снижение артериального давления, тошнота.
Решающее значение в проявлении анальгетической активности молекулы морфина имеет наличие свободной фенольной группы. Так при изменении R в положении 3 молекулы кодеина в ряду метил – этил – ацил (CH3-, C2H5-, -COCH3) наблюдается снижение обезболивающего эффекта. Вероятно, это связано с потерей стерического соответствия препарата с местом связывания.
Удаление кольца Е в структуре морфина приводит к полному исчезновению активности. Этот факт свидетельствует о важности наличия основного азота в данной структуре для проявления анальгетической активности. При удалении из структуры колец С и D, молекула преобразуется в бензоморфаны, обладающие умеренной анальгетической активностью и низким наркотическим и галлюциногенным действием.
Наиболее важными функциональными группами в структуре морфина, необходимыми для проявления анальгетического эффекта, являются
a) водород фенольного ядра, для образования водородных связей
b) и само фенольное ядро, которое участвует в Ван-дер-ваальсовых связях.
c) азот, обеспечивает ионное взаимодействие
За угнетение центров болевой чувствительности и блокирование импульсов в коре головного мозга, при воздействии на них анальгетических средств, ответственны опиатные рецепторы. Существование их в мозге показано в опытах на мышах, при исследовании стереоспецифического взаимодействия аналогов морфина – агониста леворфанола и антагониста налоксона.
Был выделен липопротеин с молекулярной массой 60 тысяч и предположено, что агонист вызывает в нем конформационные изменения, в результате которых биологическим ответом является анальгезия. Лигандами опиатных рецепторов являются нейропептиды, которые связываются с опиатным рецептором и оказывают анальгезирующее действие. Их эффект, как это вообще характерно для агонистов, блокируется антагонистами опиатов.
Есть предположение, что многие анальгетики, но не морфин, который взаимодействует с опиатными рецепторами, не связываются с самими рецепторами, а являются ингибиторами фермента, разрушающего нейропептиды, в частности пентапептиды. Таким образом, эти анальгетики повышают уровень этих нейропептидов и оказывают обезболивающий эффект. При гидролизе в мозге человека липопротеина, образуются анальгетические полипептиды – эндорфины. Наиболее активный из них β‑эндорфин вдвое активнее морфина и вызывает более длительную анальгезию, которая снимается налоксоном. Некоторые ученые рассматривают β‑эндорфин в качестве гормона, подавляющего высвобождение других нейромедиаторов.
В организме человека анальгетики начинают выделяться только как реакция на возникновение боли, т.е. их взаимодействие с опиатными рецепторами может рассматриваться как компенсаторный эффект.
Возникновение при применении морфина психической зависимости направило изыскания учеными более простых аналогов, не вызывающих эйфорию. Например, синтез промидола и лидола.
При синтезе промидола исходят из ацетона и винилацетилена, конденсация которых приводит к 2‑метилгексадиен‑2,5‑ин‑3. Далее гидратация по Кучерову дает кетон, при взаимодействии которого с метиламином образуется замещенный пиперидон. Реакция последнего с фениллитием дает замещенный пиперидол, который ацилируют пропионилхлоридом.
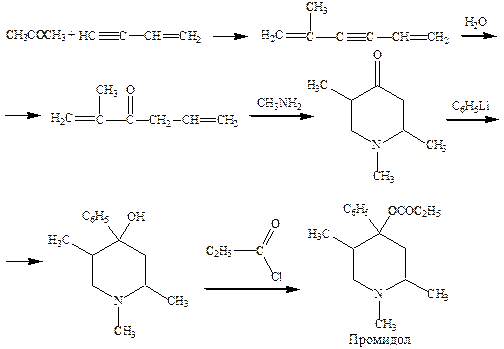
Недостаток указанного способа – получение на конечной стадии трех изомеров, вследствие чего выделение целевого продукта затрудняется.
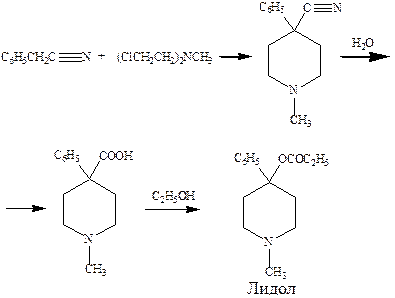
В этом синтезе лидола исходят из бензилцианида, конденсация которого с бисхлорэтилметиламином и последующим омылением продуктов конденсации нитрильной группы дают кислоту, а этерификация этой кислоты приводит к лидолу.
Характер связывания опийных анальгетиков с рецепторами существенно зависит от структурных особенностей конкретного соединения. Так полные агонисты рецепторов – морфин, промедол. Истинным антагонистом опиатных рецепторов является налоксон. Он блокирует связывание агонистов с этими рецепторами и способен вытеснить их, нарушая их рецептивные взаимодействия. Благодаря этому свойству налоксон может использоваться при интоксикации, вызванной наркотическими анальгетиками.
Однако есть и препараты обладающие смешанной активностью, давая в зависимости от типа рецепторов агонистический или антагонистический эффекты. Например, соединение с тривиальным названием налорфин вследствие агонистического эффекта оказывает анальгетический эффект, но слабее чем морфин. С другой стороны он ослабляет угнетение дыхание и снижение артериального давления, вызванное морфином.
История медицинской химии
Впервые курс медицинской химии был прочитан в МГУ в 1998 году. Как наука медицинская химия сформировалась к 1970 году.
В 19 веке медицинская химия начала прогрессировать. Были выделены средство против малярии из хинного дерева, морфин, салициловая кислота (противомикробное средство).
В 1888 году Байер впервые в мире поставил промышленное производство фенацетина
а в 1899 году – производство аспирина
К этому времени люди научились анализировать белки, жиры и углеводы, которые являются основными мишенями действия лекарственных препаратов.
В 1910 году Пауль Эрлих изобрел средство от сифилиса – сальварсан
Это обусловило возникновение концепции химиотерапии. Концепция химиотерапии предполагает не просто применение химических веществ для лечения патологий, но и необходимость модификации структур предполагаемых лекарственных соединений с тем чтобы максимально эффективно воздействовать на пораженный орган.
Эрлих также разработал теорию рецепторов и структурных изменений физиологически активных органических соединений, происходящих при их взаимодействии с рецепторами. Эта теория явилась отправной точкой для медицинской химии.
В 1928 году был открыт пенициллин, а в 1944 году Ваксберг открыл стрептомицин. Так началась эра антибиотиков.
В 60-ые годы были разработаны психотропные препараты: аминазин, мекробамат (транквилизатор).
а также резерпин, метилдофа (сердечно-сосудистые препараты).
Все эти препараты были открыты «на ощупь». Ученые продолжали искать ФАВ.
ФАВ (БАВ) – физиологически (биологически) активные вещества.
С другой стороны, проникновение компьютерных методов в органическую химию привело к бурному развитию методов расчета структуры молекул (геометрия и конформация, заряды, молекулярные орбитали, электростатические потенциалы, топологические индексы и т.д.). В силу этого количественное описание структурных особенностей даже очень сложных молекул биологического уровня становится обычным инструментом химика-органика.
В 70-ых годах была создана методологическая основа для возникновения и использования рациональных подходов к синтезу ФАВ, которая называется drug design. Это привело к формированию медицинской химии с ее современным аппаратом.
Медицинская химия – междисциплинарная наука, в ней задействованы органическая, биоорганическая, фармацевтическая химии, фармакология и биохимия. Но они не дают ответа на вопросы: «Какую структуру надо синтезировать, чтобы создать ФАВ?», «Какую структурную формулу спрогнозировать для конкретного кандидата в лекарство?». Эти вопросы являются центральными для медицинской химии.
Предметом медицинской химии является поиск и структурный дизайн ФАВ, выявление взаимосвязи химической структуры и физиологической активности и решение обратной задачи «строение-свойство», а именно конструирование необходимых структур обладающих заданным свойством.
Процесс поиска и конструирования включает в себя три основные стадии:
- Поиск соединения лидера (lead compound).
- Оптимизация соединения лидера.
- Разработка лекарственного препарата.
Стратегия поиска лекарственных препаратов зависит от накопленных знаний о препаратах, мишени, их взаимодействия и т.д. Под названием «мишень» понимают клетки, ткани, органы, функциональные системы, ферменты, акцепторы, рецепторы.
Рецепторы по Эрлиху – это небольшой участок химически определенный на большой молекуле протоплазмы, участвующей в питании и метаболизме клетки, и способный связывать специфические антигены или лекарственные вещества.
Антигены – вещества, которые воспринимаются организмом как чужеродные и вызывают специфический иммунный эффект.
Рецепторы – главным образом белковые структуры, функции которых заключаются в узнавании химического сигнала и последующей его трансформации в адекватный ответ клетки. Другими словами рецепторы представляют собой материальные субстраты активности и чувствительности клеток.
Возможны четыре ситуации при начале поиска лекарственного средства:
- Структура как рецептора, так и лиганда неизвестны. Рецептор является мишенью лекарственного вещества в организме, а понятие лиганд предполагает любое эндогенное (endon – внутри) соединение, взаимодействующее с этим рецептором в организме.
- Известна только структура рецептора.
- Известна только структура лиганда.
- Известна структура как рецептора, так и лиганда.
Поиск и конструирование соединений лидеров
Первая стадия поиска состоит в идентификации и синтезе новых ФАВ, называемых соединениями лидерами. Это структурный прототип будущего лекарства, т.е. соединение, обладающее определенной функциональной активностью, на базе которого и будет создаваться лекарство. Соединение лидер может быть найдено случайно, например, нитроглицерин (сердечно-сосудистый препарат).
Обычно начальный поиск связан с систематическим тестированием (скринингом) различных веществ на активность. Скрининг бывает двух видов:
- Исследование в одном биологическом тесте достаточно большого количества соединений.
- Изучение нескольких соединений с оригинальной структурой на многих биологических тестах.
Это дорогостоящие и сложные тесты.
Hit compound – попадание в точку, нахождение соединения, проявляющего высокую физиологическую активность («hit» - горячий).
Затем проводят тестирование круга соединений с похожей структурой, из которых потом и выбирается соединение лидер.
Таким образом, из народных средств было найдено противораковое средство - таксол.
Чаще всего соединение лидер ищут путем тотального скрининга – through-put screening.
С развитием компьютеров был разработан тотальный (сплошной) скрининг, который представляет собой одновременный автоматизированный и миниатюризированный анализ in vitro («in vitro» - вне организма, «in vivo» - внутри организма) нескольких сотен и даже тысяч соединений минимум в 50 биологических тестах.
Метод сплошного скрининга используется для тестов с замещением лигандов, меченых радиоактивными атомами и ингибированием ферментов.
Развитие методов сплошного скрининга вызвало к жизни новое направление в органическом синтезе – синтез комбинаторных библиотек. Это смесь большого числа соединений, полученных однотипным методом с использованием серии аналогичных реагентов и имеющих регулируемый состав.
Эта смесь подвергается тотальному скринингу, после чего проводится идентификация тех структур, которые проявляют биологическую активность. Очевидно, что генерируемые структуры достаточно похожи на свой прототип (так называемые терапевтические копии).
Этот подход имеет специфические аспекты, обуславливающие его применение. Например, если соединением лидером служит известное лекарство, имеющее достаточно выраженный побочный эффект, то разрабатываться будет это не основное свойство.
Дата: 2019-04-23, просмотров: 415.